
Effects of the Crystalline Properties of Hollow Ceria Nanostructures on a CuO-CeO2 Catalyst in CO Oxidation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis
2.3. Characterizations
2.4. CO Oxidation
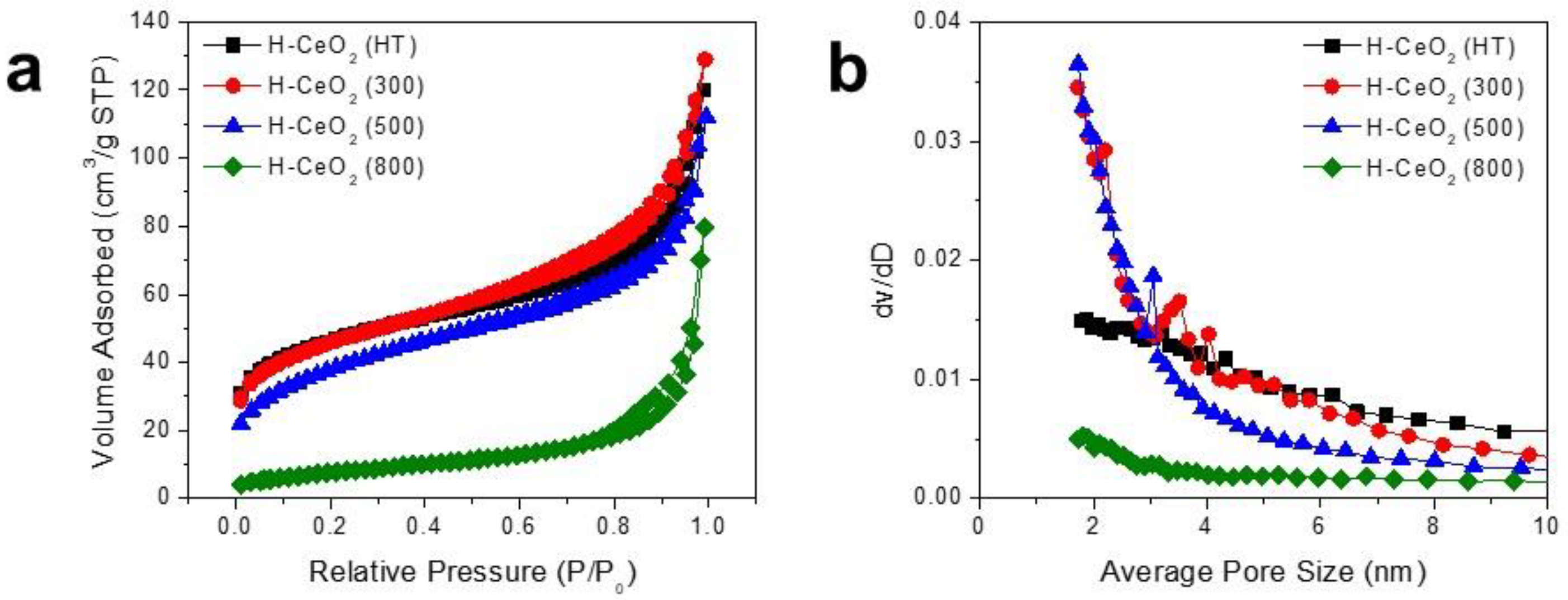
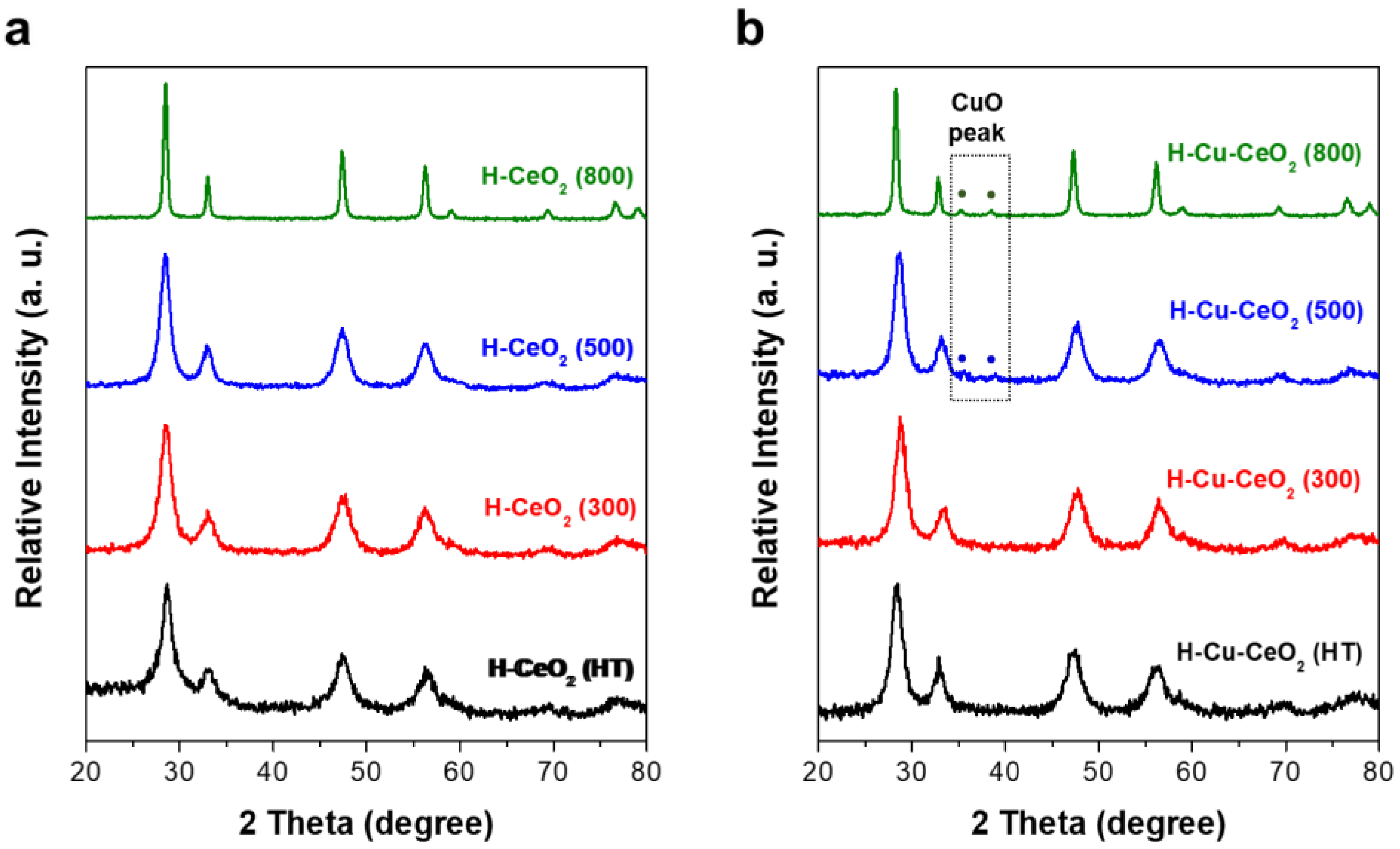
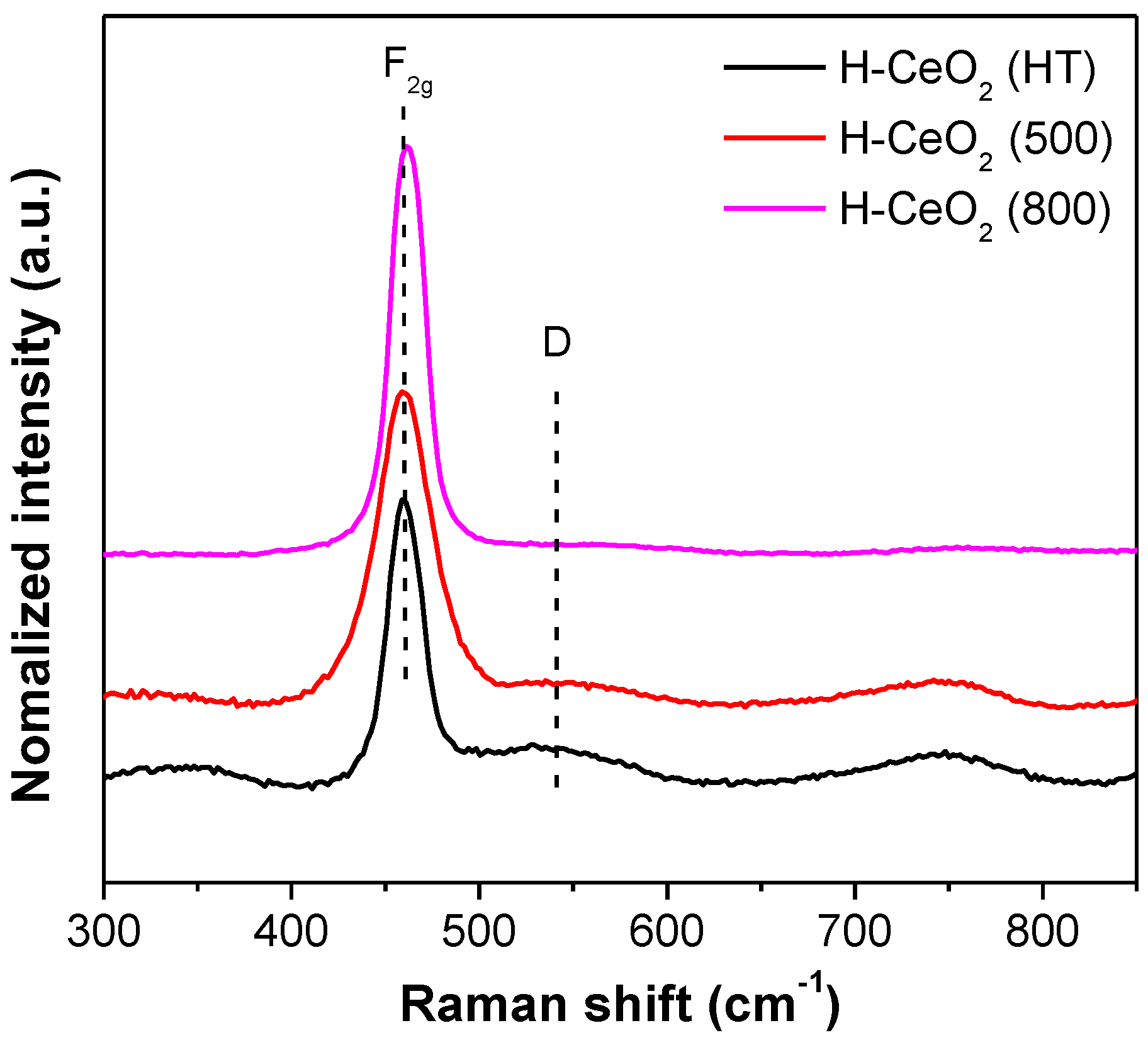
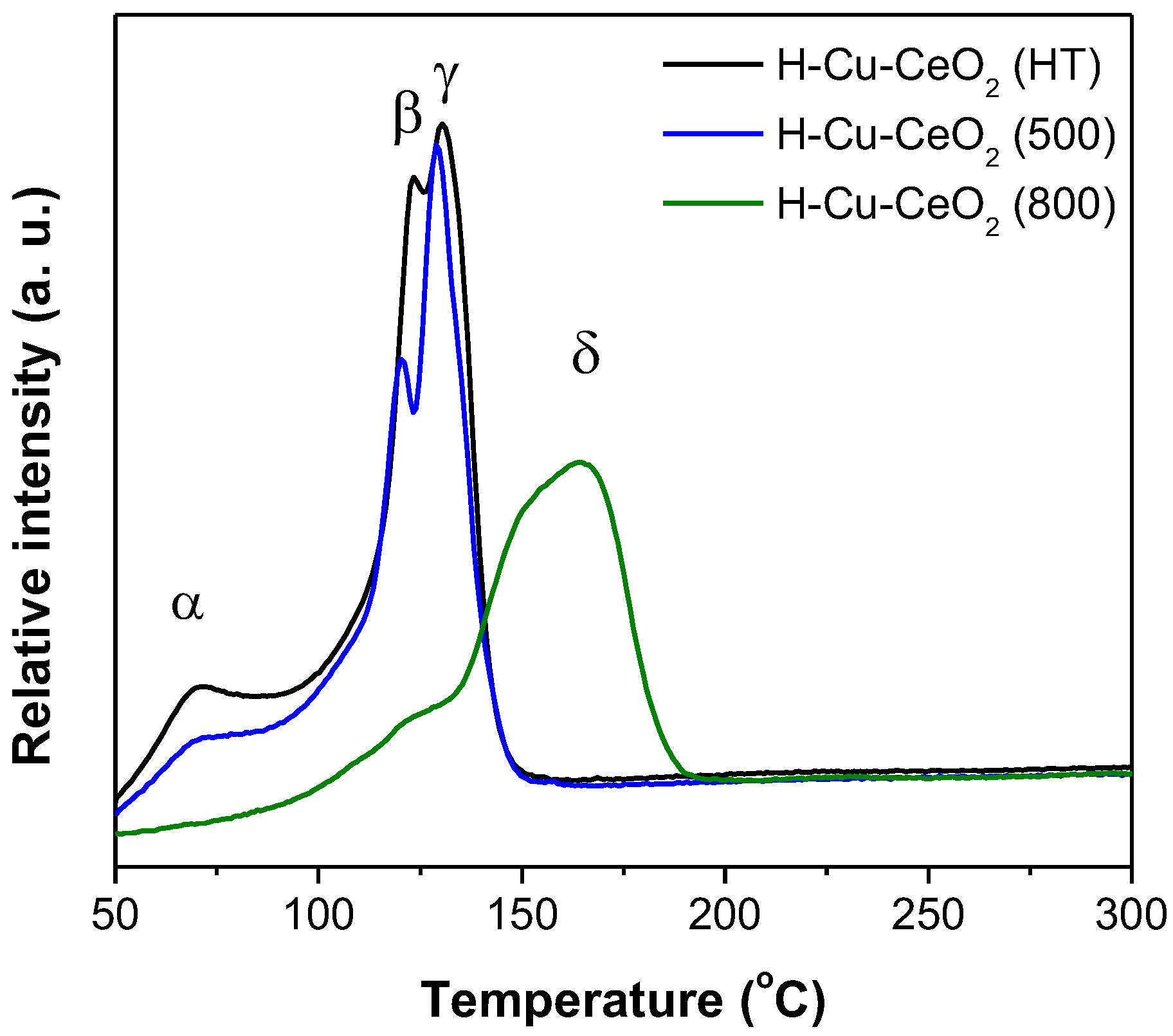
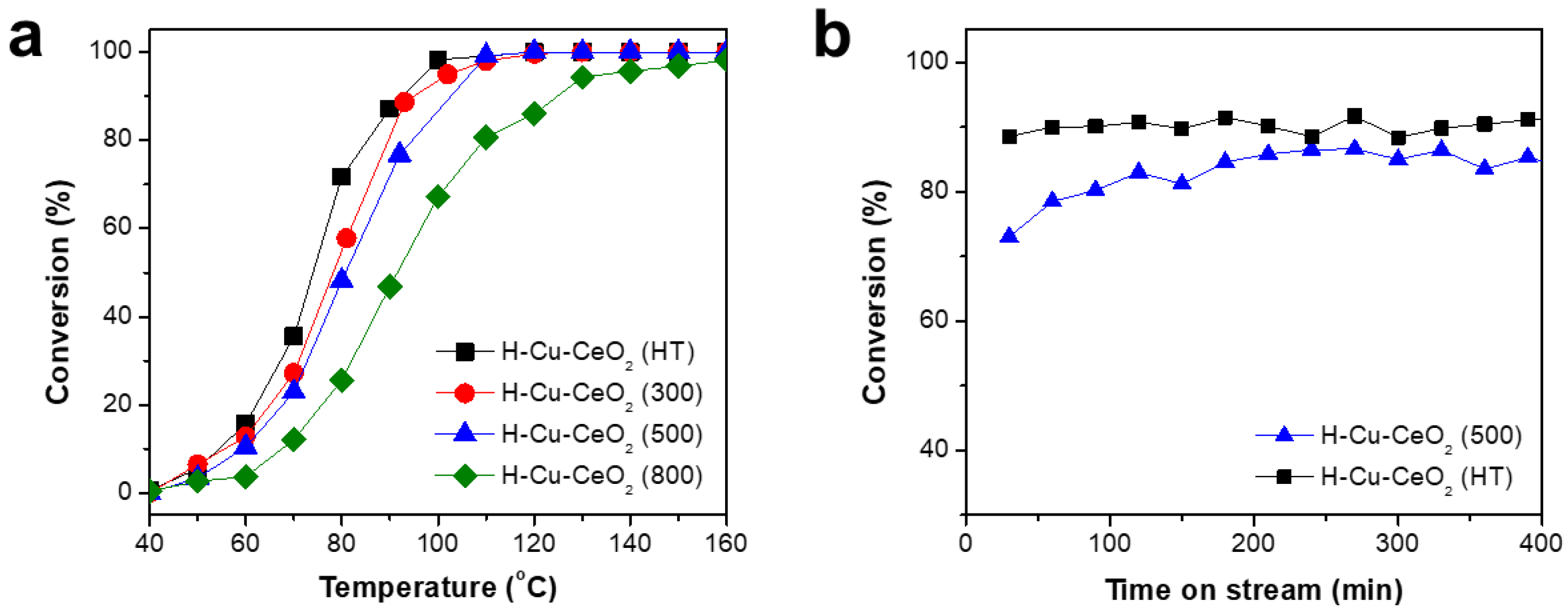
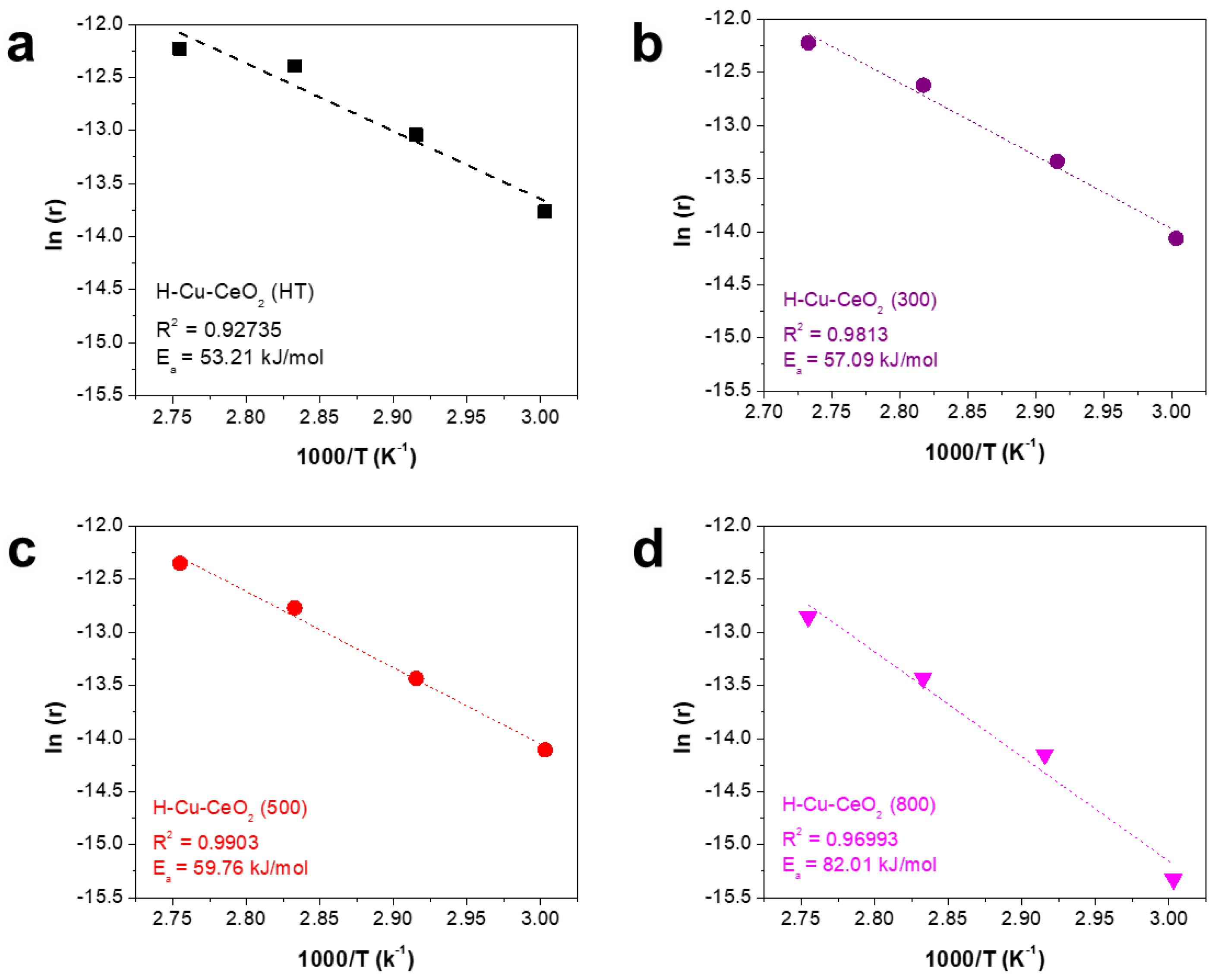
3. Results and Discussion
- (1)
- Ce3+-□-Cu+ + CO ↔ Ce3+-□-Cu+-CO
- (2)
- Ce3+- □-Cu+-CO + Ce4+-O2--Cu2+ → 2[Ce3+- □-Cu+] + CO2
- (3)
- 2[Ce3+- □-Cu+] + O2 → 2 [Ce4+-O2--Cu2+]
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Cao, A.; Wang, Z.; Li, H.; Elnabawy, A.O.; Nørskov, J.K. New insights on CO and CO2 hydrogenation for methanol synthesis: The key role of adsorbate-adsorbate interactions on Cu and the highly active MgO-Cu interface. J. Catal. 2021, 400, 325–331. [Google Scholar] [CrossRef]
- Denise, B.; Sneeden, R.P.A. Oxide-supported copper catalysts prepared from copper formate: Differences in behavior in methanol synthesis from CO/H2 and CO2/H2 mixtures. Appl. Catal. 1986, 28, 235–239. [Google Scholar] [CrossRef]
- Wolf, M.; Fischer, N.; Claeys, M. Formation of metal-support compounds in cobalt-based Fischer-Tropsch synthesis: A review. Chem Catal. 2021, 1, 1014–1041. [Google Scholar] [CrossRef]
- Toncón-Leal, C.F.; Múnera, J.F.; Arroyo-Gómez, J.J.; Sapag, K. Fe, Co and Fe/Co catalysts supported on SBA-15 for Fischer-Tropsch Synthesis. Catal. Today 2021. [Google Scholar] [CrossRef]
- Zhang, F.; Xu, X.; Qiu, Z.; Feng, B.; Liu, Y.; Xing, A.; Fan, M. Improved methanol synthesis performance of Cu/ZnO/Al2O3 catalyst by controlling its precursor structure. Green Energy Environ. 2020. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kühl, S.; Hävecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.-L.; et al. The active site of methanol synthesis over Cu/ZnO/Al2O3 Industrial Catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, H.; Qian, W.; Wu, X.; Ma, H.; Sun, Q.; Ying, W. Sodium modified Fe-Mn microsphere catalyst for Fischer–Tropsch synthesis of light olefins. Catal. Today 2022, 388, 199–207. [Google Scholar] [CrossRef]
- Ma, Z.; Zhou, C.; Wang, D.; Wang, Y.; He, W.; Tan, Y.; Liu, Q. Co-precipitated Fe-Zr catalysts for the Fischer-Tropsch synthesis of lower olefins (C2O~C4O): Synergistic effects of Fe and Zr. J. Catal. 2019, 378, 209–219. [Google Scholar] [CrossRef]
- Yeom, C.; Kim, Y. Mesoporous alumina with high capacity for carbon monoxide adsorption. Korean J. Chem. Eng. 2018, 35, 587–593. [Google Scholar] [CrossRef]
- Yeom, C.; Selvaraj, R.; Kim, Y. Preparation of nanoporous alumina using aluminum chloride via precipitation templating method for CO adsorbent. J. Ind. Eng. Chem. 2018, 67, 132–139. [Google Scholar] [CrossRef]
- Feyzbar-Khalkhali-Nejad, F.; Hassani, E.; Leonard, K.D.; Oh, T.-S. A highly stable CuO-derived adsorbent with dual Cu(I) sites for selective CO adsorption. Sep. Purif. Technol. 2022, 290, 120906. [Google Scholar] [CrossRef]
- Straczewski, G.; Vargas, C.; Zampieri, A.; Garbev, K.; Leibold, H.; Dahmen, N. Total oxidation of carbon monoxide, VOC and reduction of NO2 with catalytic ceramic filter media. Fuel Commun. 2021, 9, 100038. [Google Scholar] [CrossRef]
- Jie, W.; Liu, Y.; Deng, W.; Liu, Q.; Qiu, M.; Liu, S.; Hu, J.; Gong, L. Effect of one-dimensional ceria morphology on CuO/CeO2 catalysts for CO preferential oxidation. J. Solid State Chem. 2022, 311, 123109. [Google Scholar] [CrossRef]
- Zhang, X.; Su, L.; Kong, Y.; Ma, D.; Ran, Y.; Peng, S.; Wang, L.; Wang, Y. CeO2 nanoparticles modified by CuO nanoparticles for low-temperature CO oxidation with high catalytic activity. J. Phys. Chem. Solids 2020, 147, 109651. [Google Scholar] [CrossRef]
- Cui, X.; Zhang, X.; Yang, Z.; Xiao, M.; Ren, J.; Xiao, X.; Yude, W. The rod-like CeO2 supported by the low-loading Au nanoparticles for the efficient catalytic oxidation of CO at room temperature. Int. J. Hydrogen Energy 2022, 47, 11813–11826. [Google Scholar] [CrossRef]
- Li, Z.; Wang, H.; Zhao, W.; Xu, X.; Jin, Q.; Qi, J.; Yu, R.; Wang, D. Enhanced catalytic activity of Au-CeO2/Al2O3 monolith for low-temperature CO oxidation. Catal. Commun. 2019, 129, 105729. [Google Scholar] [CrossRef]
- Lee, K.R.; Yun, D.; Park, D.S.; Yun, Y.S.; Song, C.K.; Kim, Y.; Park, J.; Yi, J. In situmanipulation of the d-band center in metals for catalytic activity in CO oxidation. Chem. Commun. 2021, 57, 3403–3406. [Google Scholar] [CrossRef]
- Kang, M.Y.; Yun, H.J.; Yu, S.; Kim, W.; Kim, N.D.; Yi, J. Effect of TiO2 crystalline phase on CO oxidation over CuO catalysts supported on TiO2. J. Mol. Catal. A Chem. 2013, 368, 72–77. [Google Scholar] [CrossRef]
- Jiang, B.; Cha, X.; Huang, Z.; Hu, S.; Xu, K.; Cai, D.; Xiao, J.; Zhan, G. Green fabrication of hierarchically-structured Pt/bio-CeO2 nanocatalysts using natural pollen templates for low-temperature CO oxidation. Mol. Catal. 2022, 524, 112251. [Google Scholar] [CrossRef]
- Ma, K.; Liao, W.; Shi, W.; Xu, F.; Zhou, Y.; Tang, C.; Lu, J.; Shen, W.; Zhang, Z. Ceria-supported Pd catalysts with different size regimes ranging from single atoms to nanoparticles for the oxidation of CO. J. Catal. 2022, 407, 104–114. [Google Scholar] [CrossRef]
- Xu, G.; Liu, F.; Lu, Z.; Talib, S.H.; Ma, D.; Yang, Z. Design of promising single Rh atom catalyst for CO oxidation based on Graphdiyne sheets. Phys. E Low-Dimens. Syst. Nanostruct. 2021, 130, 114676. [Google Scholar] [CrossRef]
- Su, Y.-Q.; Qin, Y.-Y.; Wu, T.; Wu, D.-Y. Structure sensitivity of ceria-supported Au catalysts for CO oxidation. J. Catal. 2022, 407, 353–363. [Google Scholar] [CrossRef]
- Chen, M.; Cai, Y.; Yan, Z.; Goodman, D.W. On the origin of the unique properties of supported Au nanoparticles. J. Am. Chem. Soc. 2006, 128, 6341–6346. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Joo, J.B.; Yin, Y.; Zaera, F. A Yolk@Shell nanoarchitecture for Au/TiO2 catalysts. Angew. Chem. Int. Ed. 2011, 50, 10208–10211. [Google Scholar] [CrossRef]
- Tiscornia, I.S.; Lacoste, A.M.; Gómez, L.E.; Boix, A.V. CuO–CeO2/SiO2 coating on ceramic monolith: Effect of the nature of the catalyst support on CO preferential oxidation in a H2-rich stream. Int. J. Hydrogen Energy 2020, 45, 6636–6650. [Google Scholar] [CrossRef]
- Papavasiliou, A.; Van Everbroeck, T.; Blonda, C.; Oliani, B.; Sakellis, E.; Cool, P.; Canu, P.; Katsaros, F.K. Mesoporous CuO/TiO2 catalysts prepared by the ammonia driven deposition precipitation method for CO preferential oxidation: Effect of metal loading. Fuel 2022, 311, 122491. [Google Scholar] [CrossRef]
- Shi, W.; Gao, T.; Zhang, L.; Ma, Y.; Liu, Z.; Zhang, B. Tailoring the surface structures of iron oxide nanorods to support Au nanoparticles for CO oxidation. Chin. J. Catal. 2019, 40, 1884–1894. [Google Scholar] [CrossRef]
- Tang, H.; Liu, F.; Wei, J.; Qiao, B.; Zhao, K.; Su, Y.; Jin, C.; Li, L.; Liu, J.J.; Wang, J.; et al. Ultrastable hydroxyapatite/titanium-dioxide-supported gold nanocatalyst with strong metal–support interaction for carbon monoxide oxidation. Angew. Chem. Int. Ed. 2016, 55, 10606–10611. [Google Scholar] [CrossRef]
- Zhang, X.-m.; Tian, P.; Tu, W.; Zhang, Z.; Xu, J.; Han, Y.-F. Tuning the dynamic interfacial structure of copper–ceria catalysts by indium oxide during CO oxidation. ACS Catal. 2018, 8, 5261–5275. [Google Scholar] [CrossRef]
- Yao, X.; Gao, F.; Yu, Q.; Qi, L.; Tang, C.; Dong, L.; Chen, Y. NO reduction by CO over CuO–CeO2 catalysts: Effect of preparation methods. Catal. Sci. Technol. 2013, 3, 1355–1366. [Google Scholar] [CrossRef]
- Zabilskiy, M.; Djinović, P.; Tchernychova, E.; Tkachenko, O.P.; Kustov, L.M.; Pintar, A. Nanoshaped CuO/CeO2 materials: Effect of the exposed ceria surfaces on catalytic activity in N2O decomposition reaction. ACS Catal. 2015, 5, 5357–5365. [Google Scholar] [CrossRef]
- May, Y.A.; Wang, W.-W.; Yan, H.; Wei, S.; Jia, C.-J. Insights into facet-dependent reactivity of CuO–CeO2 nanocubes and nanorods as catalysts for CO oxidation reaction. Chin. J. Catal. 2020, 41, 1017–1027. [Google Scholar] [CrossRef]
- Maciel, C.G.; Silva, T.d.F.; Hirooka, M.I.; Belgacem, M.N.; Assaf, J.M. Effect of nature of ceria support in CuO/CeO2 catalyst for PROX-CO reaction. Fuel 2012, 97, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Jang, H.S.; Kim, N.Y.; Joo, J.B. Cu-doped TiO2 hollow nanostructures for the enhanced photocatalysis under visible light conditions. J. Ind. Eng. Chem. 2021, 99, 352–363. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Lee, H.; Choi, D.-S.; Kim, J.; Jang, H.-S.; Kim, N.-Y.; Joo, J.-B. Synthesis of hollow mesoporous TiN nanostructures as an efficient catalyst support for methanol electro-oxidation. Catalysts 2021, 11, 763. [Google Scholar] [CrossRef]
- Choi, I.; Lee, H.K.; Lee, G.W.; Kim, J.; Joo, J.B. Inorganic shell nanostructures to enhance performance and stability of metal nanoparticles in catalytic applications. Rare Metals 2020, 39, 767–783. [Google Scholar] [CrossRef]
- Joo, J.B.; Vu, A.; Zhang, Q.; Dahl, M.; Gu, M.; Zaera, F.; Yin, Y. A Sulfated ZrO2 hollow nanostructure as an acid catalyst in the dehydration of fructose to 5-hydroxymethylfurfural. ChemSusChem 2013, 6, 2001–2008. [Google Scholar] [CrossRef]
- Joo, J.B.; Dahl, M.; Li, N.; Zaera, F.; Yin, Y. Tailored synthesis of mesoporous TiO2 hollow nanostructures for catalytic applications. Energy Environ. Sci. 2013, 6, 2082–2092. [Google Scholar] [CrossRef]
- Joo, J.B.; Lee, I.; Dahl, M.; Moon, G.D.; Zaera, F.; Yin, Y. Controllable synthesis of mesoporous TiO2 hollow shells: Toward an efficient photocatalyst. Adv. Funct. Mater. 2013, 23, 4246–4254. [Google Scholar] [CrossRef]
- Dillon, R.J.; Joo, J.-B.; Zaera, F.; Yin, Y.; Bardeen, C.J. Correlating the excited state relaxation dynamics as measured by photoluminescence and transient absorption with the photocatalytic activity of Au@TiO2 core–shell nanostructures. Phys. Chem. Chem. Phys. 2013, 15, 1488–1496. [Google Scholar] [CrossRef]
- Joo, J.B.; Zhang, Q.; Dahl, M.; Lee, I.; Goebl, J.; Zaera, F.; Yin, Y. Control of the nanoscale crystallinity in mesoporous TiO2 shells for enhanced photocatalytic activity. Energy Environ. Sci. 2012, 5, 6321–6327. [Google Scholar] [CrossRef]
- Al-Marri, A.H.; Khan, M.; Shaik, M.R.; Mohri, N.; Adil, S.F.; Kuniyil, M.; Alkhathlan, H.Z.; Al-Warthan, A.; Tremel, W.; Tahir, M.N.; et al. Green synthesis of Pd@graphene nanocomposite: Catalyst for the selective oxidation of alcohols. Arab. J. Chem. 2016, 9, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Adil, S.F.; Assal, M.E.; Shaik, M.R.; Kuniyil, M.; AlOtaibi, N.M.; Khan, M.; Sharif, M.; Alam, M.M.; Al-Warthan, A.; Mohammed, J.A.; et al. A Facile synthesis of ZrOx-MnCO3/graphene oxide (GRO) nanocomposites for the oxidation of alcohols using molecular oxygen under base free conditions. Catalysts 2019, 9, 759. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Wu, J.; Zhang, A.; Xue, L.; Wang, Q.; Tian, X.; Shan, S.; Zhong, C.-J.; Zeng, S. Hollow copper–ceria microspheres with single and multiple shells for preferential CO oxidation. CrystEngComm 2019, 21, 3619–3626. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Y.; Zhou, Y.; Xiang, S.; Wang, Q.; Zhang, C.; Sheng, X. CeO2 hollow nanospheres synthesized by a one pot template-free hydrothermal method and their application as catalyst support. RSC Adv. 2015, 5, 58237–58245. [Google Scholar] [CrossRef]
- Lykaki, M.; Pachatouridou, E.; Carabineiro, S.A.C.; Iliopoulou, E.; Andriopoulou, C.; Kallithrakas-Kontos, N.; Boghosian, S.; Konsolakis, M. Ceria nanoparticles shape effects on the structural defects and surface chemistry: Implications in CO oxidation by Cu/CeO2 catalysts. Appl. Catal. B Environ. 2018, 230, 18–28. [Google Scholar] [CrossRef]
- Ye, R.-P.; Li, Q.; Gong, W.; Wang, T.; Razink, J.J.; Lin, L.; Qin, Y.-Y.; Zhou, Z.; Adidharma, H.; Tang, J.; et al. High-performance of nanostructured Ni/CeO2 catalyst on CO2 methanation. Appl. Catal. B Environ. 2020, 268, 118474. [Google Scholar] [CrossRef]
- Das, S.; Ashok, J.; Bian, Z.; Dewangan, N.; Wai, M.H.; Du, Y.; Borgna, A.; Hidajat, K.; Kawi, S. Silica–Ceria sandwiched Ni core–shell catalyst for low temperature dry reforming of biogas: Coke resistance and mechanistic insights. Appl. Catal. B Environ. 2018, 230, 220–236. [Google Scholar] [CrossRef]
- Jang, W.-J.; Kim, H.-M.; Shim, J.-O.; Yoo, S.-Y.; Jeon, K.-W.; Na, H.-S.; Lee, Y.-L.; Jeong, D.-W.; Bae, J.W.; Nah, I.W.; et al. Key properties of Ni–MgO–CeO2, Ni–MgO–ZrO2, and Ni–MgO–Ce(1−x)Zr(x)O2 catalysts for the reforming of methane with carbon dioxide. Green Chem. 2018, 20, 1621–1633. [Google Scholar] [CrossRef]
- He, D.; Hao, H.; Chen, D.; Liu, J.; Yu, J.; Lu, J.; Liu, F.; Wan, G.; He, S.; Luo, Y. Synthesis and application of rare-earth elements (Gd, Sm, and Nd) doped ceria-based solid solutions for methyl mercaptan catalytic decomposition. Catal. Today 2017, 281, 559–565. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Ioannides, T. Effect of synthesis parameters on catalytic properties of CuO-CeO2. Appl. Catal. B Environ. 2006, 67, 1–11. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, S.-J.; Lee, H.; Kim, J.; Kim, N.-Y.; Choi, D.-S.; Joo, J.B. Effects of the Crystalline Properties of Hollow Ceria Nanostructures on a CuO-CeO2 Catalyst in CO Oxidation. Materials 2022, 15, 3859. https://doi.org/10.3390/ma15113859
Jang S-J, Lee H, Kim J, Kim N-Y, Choi D-S, Joo JB. Effects of the Crystalline Properties of Hollow Ceria Nanostructures on a CuO-CeO2 Catalyst in CO Oxidation. Materials. 2022; 15(11):3859. https://doi.org/10.3390/ma15113859
Chicago/Turabian StyleJang, Se-Jin, Hyeonkyeong Lee, Jiyull Kim, Na-Yeon Kim, Dong-Seop Choi, and Ji Bong Joo. 2022. "Effects of the Crystalline Properties of Hollow Ceria Nanostructures on a CuO-CeO2 Catalyst in CO Oxidation" Materials 15, no. 11: 3859. https://doi.org/10.3390/ma15113859