
Evolution of Topological Surface States Following Sb Layer Adsorption on Bi2Se3


Abstract
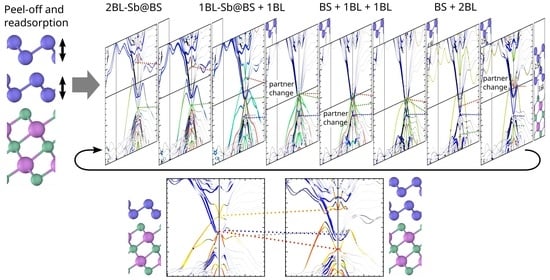
:
1. Introduction
2. Results
2.1. Structure of Bulk
2.2. Structure of Bulk Sb and Isolated Sb Sheets
2.3. Structural Details of the Sb@BS System
2.4. Band Structures
- The topological surface states “TSSs” which form at the interface between the TI and the CI.
- The Dirac point “D*”, which is the crossing of the two TSSs.
- The peak “” which is derived from in the Sb atoms.
- The band “B” which “connects” the TSS with the Sb peak.
- The bands “” which stem from a Rashba splitting of the BS conduction bands.
2.5. Band Hybridization
3. Discussion
3.1. Structural Details
3.2. Band Structures
4. Materials and Methods
4.1. Computational Parameters
4.2. Cell Optimization and Supercells
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vobornik, I.; Manju, U.; Fujii, J.; Borgatti, F.; Torelli, P.; Krizmancic, D.; Hor, Y.S.; Cava, R.J.; Panaccione, G. Magnetic Proximity Effect as a Pathway to Spintronic Applications of Topological Insulators. Nano Lett. 2011, 11, 4079–4082. [Google Scholar] [CrossRef]
- Hutasoit, J.A.; Stanescu, T.D. Induced spin texture in semiconductor/topological insulator heterostructures. Phys. Rev. B 2011, 84, 085103. [Google Scholar] [CrossRef]
- Maier, L.; Oostinga, J.B.; Knott, D.; Brüne, C.; Virtanen, P.; Tkachov, G.; Hankiewicz, E.M.; Gould, C.; Buhmann, H.; Molenkamp, L.W. Induced Superconductivity in the Three-Dimensional Topological Insulator HgTe. Phys. Rev. Lett. 2012, 109, 186806. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Katmis, F.; Assaf, B.A.; Steinberg, H.; Jarillo-Herrero, P.; Heiman, D.; Moodera, J.S. Exchange-Coupling-Induced Symmetry Breaking in Topological Insulators. Phys. Rev. Lett. 2013, 110, 186807. [Google Scholar] [CrossRef] [PubMed]
- Eremeev, S.V.; Men’shov, V.N.; Tugushev, V.V.; Echenique, P.M.; Chulkov, E.V. Magnetic proximity effect at the three-dimensional topological insulator/magnetic insulator interface. Phys. Rev. B 2013, 88, 144430. [Google Scholar] [CrossRef]
- Xu, J.P.; Liu, C.; Wang, M.X.; Ge, J.; Liu, Z.L.; Yang, X.; Chen, Y.; Liu, Y.; Xu, Z.A.; Gao, C.L.; et al. Artificial Topological Superconductor by the Proximity Effect. Phys. Rev. Lett. 2014, 112, 217001. [Google Scholar] [CrossRef]
- Nadj-Perge, S.; Drozdov, I.K.; Li, J.; Chen, H.; Jeon, S.; Seo, J.; MacDonald, A.H.; Bernevig, B.A.; Yazdani, A. Observation of Majorana fermions in ferromagnetic atomic chains on a superconductor. Science 2014, 346, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Menshchikova, T.V.; Otrokov, M.M.; Tsirkin, S.S.; Samorokov, D.A.; Bebneva, V.V.; Ernst, A.; Kuznetsov, V.M.; Chulkov, E.V. Band Structure Engineering in Topological Insulator Based Heterostructures. Nano Lett. 2013, 13, 6064–6069. [Google Scholar] [CrossRef] [PubMed]
- Popov, I.; Mantega, M.; Narayan, A.; Sanvito, S. Proximity-induced topological state in graphene. Phys. Rev. B 2014, 90, 035418. [Google Scholar] [CrossRef]
- Essert, S.; Krueckl, V.; Richter, K. Using topological insulator proximity to generate perfectly conducting channels in materials without topological protection. New J. Phys. 2014, 16, 113058. [Google Scholar] [CrossRef]
- Men’shov, V.N.; Tugushev, V.V.; Eremeev, S.V.; Echenique, P.M.; Chulkov, E.V. Band bending driven evolution of the bound electron states at the interface between a three-dimensional topological insulator and a three-dimensional normal insulator. Phys. Rev. B 2015, 91, 075307. [Google Scholar] [CrossRef]
- Seixas, L.; West, D.; Fazzio, A.; Zhang, S.B. Vertical twinning of the Dirac cone at the interface between topological insulators and semiconductors. Nat. Commun. 2015, 6, 7630. [Google Scholar] [CrossRef]
- Hirahara, T.; Eremeev, S.V.; Shirasawa, T.; Okuyama, Y.; Kubo, T.; Nakanishi, R.; Akiyama, R.; Takayama, A.; Hajiri, T.; Ideta, S.i.; et al. Large-Gap Magnetic Topological Heterostructure Formed by Subsurface Incorporation of a Ferromagnetic Layer. Nano Lett. 2017, 17, 3493–3500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, C.X.; Qi, X.L.; Dai, X.; Fang, Z.; Zhang, S.C. Topological insulators in Bi2Se3, Bi2Te3 and Sb2Te3 with a single Dirac cone on the surface. Nat. Phys. 2009, 5, 438–442. [Google Scholar] [CrossRef]
- Yazyev, O.V.; Moore, J.E.; Louie, S.G. Spin Polarization and Transport of Surface States in the Topological Insulators Bi2Se3 and Bi2Te3 from First Principles. Phys. Rev. Lett. 2010, 105, 266806. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Pandey, R.; Karna, S.P. Atomically Thin Group V Elemental Films: Theoretical Investigations of Antimonene Allotropes. ACS Appl. Mater. Interfaces 2015, 7, 11490–11496. [Google Scholar] [CrossRef] [PubMed]
- Aktürk, O.Ü.; Özçelik, V.O.; Ciraci, S. Single-layer crystalline phases of antimony: Antimonenes. Phys. Rev. B 2015, 91, 235446. [Google Scholar] [CrossRef]
- Flammini, R.; Colonna, S.; Hogan, C.; Mahatha, S.K.; Papagno, M.; Barla, A.; Sheverdyaeva, P.M.; Moras, P.; Aliev, Z.S.; Babanly, M.B.; et al. Evidence of β -antimonene at the Sb/Bi 2 Se 3 interface. Nanotechnology 2018, 29, 065704. [Google Scholar] [CrossRef]
- Kim, S.H.; Jin, K.H.; Kho, B.W.; Park, B.G.; Liu, F.; Kim, J.S.; Yeom, H.W. Atomically Abrupt Topological p–n Junction. ACS Nano 2017, 11, 9671–9677. [Google Scholar] [CrossRef]
- Hogan, C.; Holtgrewe, K.; Ronci, F.; Colonna, S.; Sanna, S.; Moras, P.; Sheverdyaeva, P.M.; Mahatha, S.; Papagno, M.; Aliev, Z.S.; et al. Temperature Driven Phase Transition at the Antimonene/Bi2Se3 van der Waals Heterostructure. ACS Nano 2019, 13, 10481–10489. [Google Scholar] [CrossRef]
- Shi, Z.Q.; Li, H.; Yuan, Q.Q.; Xue, C.L.; Xu, Y.J.; Lv, Y.Y.; Jia, Z.Y.; Chen, Y.; Zhu, W.; Li, S.C. Kinetics-Limited Two-Step Growth of van der Waals Puckered Honeycomb Sb Monolayer. ACS Nano 2020, 14, 16755–16760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, Z.; Duan, W.; Liu, F.; Wu, J. Topological and electronic transitions in a Sb(111) nanofilm: The interplay between quantum confinement and surface effect. Phys. Rev. B 2012, 85, 201410. [Google Scholar] [CrossRef]
- Jin, K.H.; Yeom, H.W.; Jhi, S.H. Band structure engineering of topological insulator heterojunctions. Phys. Rev. B 2016, 93, 075308. [Google Scholar] [CrossRef]
- Holtgrewe, K.; Mahatha, S.K.; Sheverdyaeva, P.M.; Moras, P.; Flammini, R.; Colonna, S.; Ronci, F.; Papagno, M.; Barla, A.; Petaccia, L.; et al. Topologization of β-antimonene on Bi2Se3 via proximity effects. Sci. Rep. 2020, 10, 14619. [Google Scholar] [CrossRef]
- García, Á.M.; del Corro, E.; Kalbac, M.; Frank, O. Tuning the electronic properties of monolayer and bilayer transition metal dichalcogenide compounds under direct out-of-plane compression. Phys. Chem. Chem. Phys. 2017, 19, 13333–13340. [Google Scholar] [CrossRef]
- Nakajima, S. The crystal structure of Bi2Te3-xSex. J. Phys. Chem. Solids 1963, 24, 479–485. [Google Scholar] [CrossRef]
- Sanna, S.; Schmidt, W.G. Density-functional theory calculations of the LiNbO3 (2-1-10), (1-100), and (0001) surfaces, commonly referred to as X, Y, and Z cuts, are presented. In case of the Z cut, we find a pronounced dependence of the surface structure and stoichiometry on the d. Phys. Rev. B 2010, 81, 214116. [Google Scholar] [CrossRef]
- Fu, L.; Kane, C.L. Time reversal polarization and a Z2 adiabatic spin pump. Phys. Rev. B 2006, 74, 195312. [Google Scholar] [CrossRef]
- Fu, L.; Kane, C.L. Topological insulators with inversion symmetry. Phys. Rev. B 2007, 76, 045302. [Google Scholar] [CrossRef]
- Yazyev, O.V.; Kioupakis, E.; Moore, J.E.; Louie, S.G. Quasiparticle effects in the bulk and surface-state bands of Bi2Se3 and Bi2Te3 topological insulators. Phys. Rev. B 2012, 85, 161101. [Google Scholar] [CrossRef]
- Nechaev, I.A.; Hatch, R.C.; Bianchi, M.; Guan, D.; Friedrich, C.; Aguilera, I.; Mi, J.L.; Iversen, B.B.; Blügel, S.; Hofmann, P.; et al. Evidence for a direct band gap in the topological insulator Bi2Se3 from theory and experiment. Phys. Rev. B 2013, 87, 121111. [Google Scholar] [CrossRef]
- Aguilera, I.; Friedrich, C.; Blügel, S. Spin-orbit coupling in quasiparticle studies of topological insulators. Phys. Rev. B 2013, 88, 165136. [Google Scholar] [CrossRef]
- Aguilera, I.; Friedrich, C.; Bihlmayer, G.; Blügel, S. GW study of topological insulators Bi2Se3,Bi2Te3, and Sb2Te3: Beyond the perturbative one-shot approach. Phys. Rev. B 2013, 88, 045206. [Google Scholar] [CrossRef]
- Crowley, J.M.; Tahir-Kheli, J.; Goddard, W.A. Accurate Ab Initio Quantum Mechanics Simulations of Bi2Se3 and Bi2Te3 Topological Insulator Surfaces. J. Phys. Chem. Lett. 2015, 6, 3792–3796. [Google Scholar] [CrossRef] [PubMed]
- Förster, T.; Krüger, P.; Rohlfing, M. Two-dimensional topological phases and electronic spectrum of Bi2Se3 thin films from GW calculations. Phys. Rev. B 2015, 92, 201404. [Google Scholar] [CrossRef]
- Förster, T.; Krüger, P.; Rohlfing, M. GW calculations for Bi2Te3 and Sb2Te3 thin films: Electronic and topological properties. Phys. Rev. B 2016, 93, 205442. [Google Scholar] [CrossRef]
- Lawal, A.; Shaari, A.; Ahmed, R.; Jarkoni, N. First-principles many-body comparative study of Bi2Se3 crystal: A promising candidate for broadband photodetector. Phys. Lett. A 2017, 381. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Feynman, R.P. Forces in Molecules. Phys. Rev. 1939, 56, 340–343. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Murnaghan, F.D. The Compressibility of Media under Extreme Pressures. Proc. Natl. Acad. Sci. USA 1944, 30, 244–247. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [Å] | [Å] | V [Å3] | u | v | [Å] | [Å] | |
|---|---|---|---|---|---|---|---|
| Experiment | 4.143 | 28.636 | 425.67 | 0.4008 | 0.2117 | 2.579 | 6.966 |
| Calculation | 4.13 | 28.73 | 425.26 | 0.4000 | 0.2111 | 2.56 | 7.02 |
| Deviation | 0.22% | −0.34% | −0.55% | 0.19% | 0.26% | 0.89% | −0.79% |
| System | Abbreviation | [meV] | [Å] | [Å] |
|---|---|---|---|---|
| Bulk (natural stacking) | b-nat | 0 | 4.31 | 2.16 |
| Bulk (matched to BS) | b-str | 46 | 4.13 | 2.16 |
| Bulk (inverted stacking) | b-inv | 40 | 4.12 | 2.84 |
| Bulk (distorted stacking) | b-dis | 47 | 4.06 | 2.94 |
| Free-standing 1BL sheet | f1BL | 276 | 4.05 | — |
| Free-standing 2BL sheet (natural stacking) | f2BL-nat | 178 | 4.16 | 2.43 |
| Free-standing 2BL sheet (inverted stacking) | f2BL-inv | 173 | 4.08 | 2.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holtgrewe, K.; Hogan, C.; Sanna, S. Evolution of Topological Surface States Following Sb Layer Adsorption on Bi2Se3. Materials 2021, 14, 1763. https://doi.org/10.3390/ma14071763
Holtgrewe K, Hogan C, Sanna S. Evolution of Topological Surface States Following Sb Layer Adsorption on Bi2Se3. Materials. 2021; 14(7):1763. https://doi.org/10.3390/ma14071763
Chicago/Turabian StyleHoltgrewe, Kris, Conor Hogan, and Simone Sanna. 2021. "Evolution of Topological Surface States Following Sb Layer Adsorption on Bi2Se3" Materials 14, no. 7: 1763. https://doi.org/10.3390/ma14071763