
First Principle Study of TiB2 (0001)/γ-Fe (111) Interfacial Strength and Heterogeneous Nucleation

Abstract
:
1. Introduction
2. Experimental and Computational Procedures
3. Results and Discussion
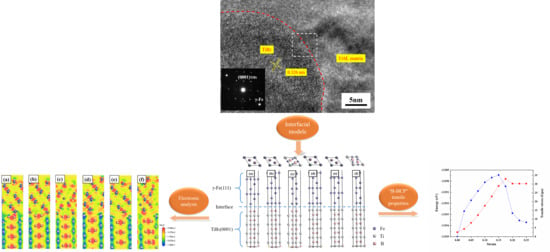
3.1. Experiment
3.2. Calculation and Simulation
3.2.1. Bulk and Surface Properties
- Bulk Properties
- Surface Energy
3.2.2. Properties of the TiB2/γ-Fe Interface
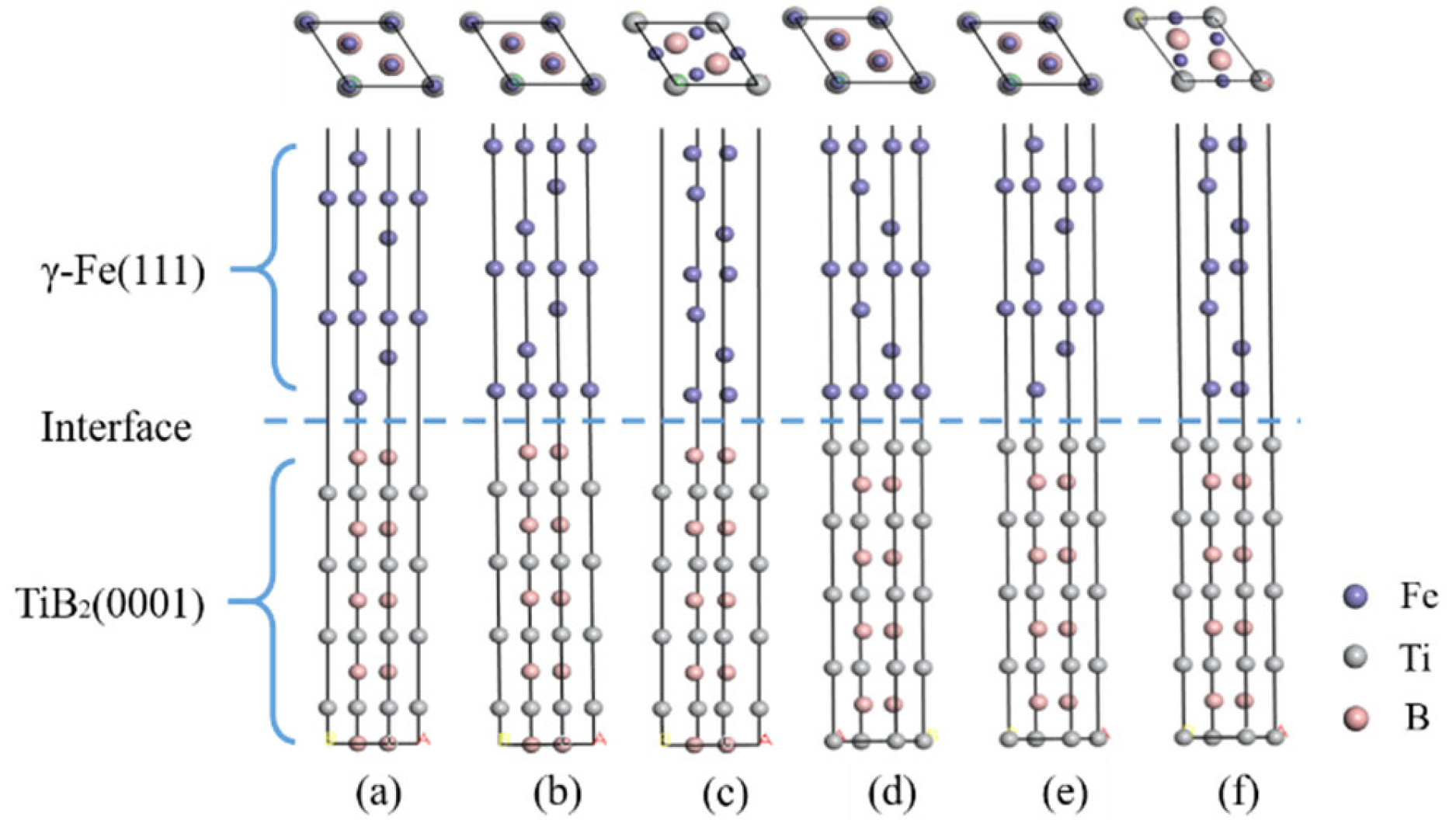
- TiB2 (0001) and γ-Fe (111) Interface
- Adhesion Work
- Interfacial Stability
- Electronic Structure and Bonding
3.2.3. Tensile Strength and Property
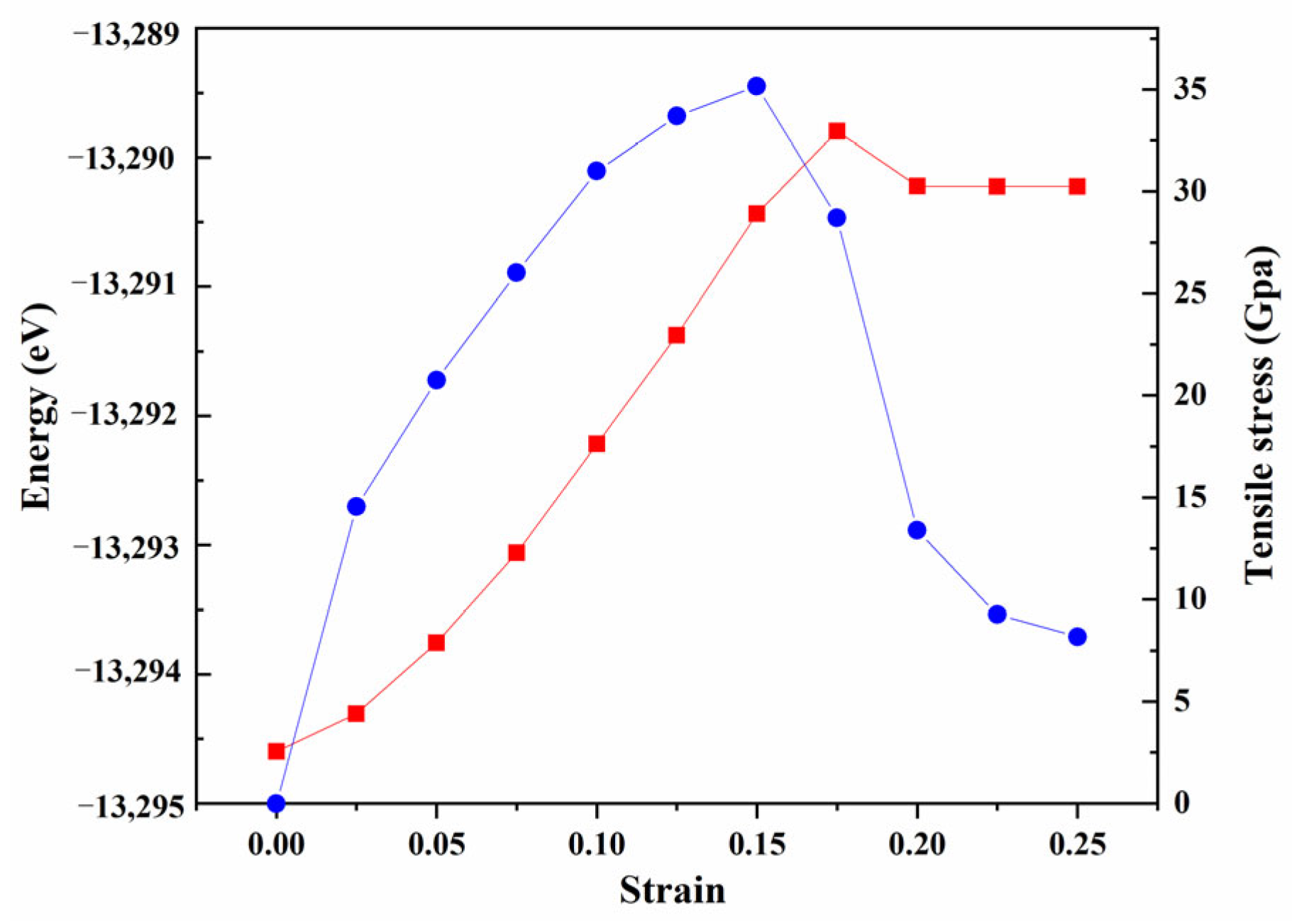
- Tensile Simulation
- Electronic property
4. Analysis on TiB2 as Heterogeneous Nucleation of γ-Fe
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Almangour, B.; Grzesiak, D.; Borkar, T.; Yang, J. Densification behavior, microstructural evolution, and mechanical properties of TiC/316L stainless steel nanocomposites fabricated by selective laser melting. Mater Des. 2018, 138, 119–128. [Google Scholar] [CrossRef]
- Almangour, B.; Grzesiak, D.; Yang, J. Rapid fabrication of bulk-form TiB2/316L stainless steel nanocomposites with novel reinforcement architecture and improved performance by selective laser melting. J. Alloys Compd. 2016, 680, 480–493. [Google Scholar] [CrossRef]
- Olsson, C.O.A.; Landolt, D. Passive films on stainless steels—Chemistry, structure and growth. Electrochim. Acta 2003, 48, 1093–1104. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, Z.; Bai, P.; Du, W.; Li, Y.; Yang, X.; Wang, Q. In-situ synthesis of TiC/graphene/Ti6Al4V composite coating by laser cladding. Mater. Lett. 2020, 270, 127711. [Google Scholar] [CrossRef]
- Abenojar, J.; Velasco, F.; Torralba, J.; Bas, J.; Calero, J.; Marcè, R. Reinforcing 316L stainless steel with intermetallic and carbide particles. Mater. Sci. Eng. A 2002, 335, 1–5. [Google Scholar] [CrossRef]
- Patankar, S.; Tan, M. Role of reinforcement in sintering of SiC/316L stainless steel composite. Powder Metall. 2013, 43, 350–352. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, J.; Bai, P.; Qu, H.; Liang, M. Microstructure and Mechanical Properties of TiC-Reinforced 316L Stainless Steel Composites Fabricated Using Selective Laser Melting. Metals 2019, 9, 267. [Google Scholar] [CrossRef] [Green Version]
- Das, M.; Bhattacharya, K.; Dittrick, S.; Mandal, C.; Balla, V.; Sampath Kumar, T.; Bandyopadhyay, A.; Manna, I. In situ synthesized TiB–TiN reinforced Ti6Al4V alloy composite coatings: Microstructure, tribological and in-vitro biocompatibility. J. Mech. Behav. Biomed. 2014, 29, 259–271. [Google Scholar] [CrossRef]
- Niu, L.; Hojamberdiev, M.; Xu, Y. Preparation of in situ-formed WC/Fe composite on gray cast iron substrate by a centrifugal casting process. J. Mater. Process. Tech. 2010, 210, 1986–1990. [Google Scholar] [CrossRef]
- Wang, Q.; Zhao, Z.; Bai, P.; Du, W.; Liao, H.; Li, Y.; Liang, M.; Huo, P.; Zhang, L.; Tie, D. Effects of alloying elements X (Cr, Mn, Mo, Ni, Si) on the interface stability of TiC (001)/γ-Fe (001) in TiC/316L stainless steel composite formed by selective laser melting: First principles and experiments. Adv. Compos. Hybrid Mater. 2021. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, L.; Bai, P.; Du, W.; Wang, S.; Xu, X.; Dong, Q.; Li, Y.; Han, B. Tribological Behavior of In Situ TiC/Graphene/Graphite/Ti6Al4V Matrix Composite Through Laser Cladding. Acta Metall. Sin. Engl. Lett. 2021. [Google Scholar] [CrossRef]
- Li, B.; Liu, Y.; Li, J.; Cao, H.; He, L. Effect of sintering process on the microstructures and properties of in situ TiB2–TiC reinforced steel matrix composites produced by spark plasma sintering. J. Mater. Process. Tech. 2010, 210, 91–95. [Google Scholar] [CrossRef]
- Antoni-Zdziobek, A.; Gospodinova, M.; Bonnet, F.; Hodaj, F. Solidification paths in the iron-rich part of the Fe–Ti–B ternary system. J. Alloys Compd. 2016, 657, 302–312. [Google Scholar] [CrossRef]
- Akhtar, F. Ceramic reinforced high modulus steel composites: Processing, microstructure and properties. Can. Metall. Quart. 2014, 53, 253–263. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, Y.; Han, H.; Li, B. Effect of Mg–TiB2 master alloy on the grain refinement of AZ91D magnesium alloy. J. Alloys Compd. 2009, 487, 202–205. [Google Scholar] [CrossRef]
- Liu, R.; Yin, X.; Feng, K.; Xu, R. First-principles calculations on Mg/TiB2 interfaces. Comp. Mater. Sci. 2018, 149, 373–378. [Google Scholar] [CrossRef]
- Deng, C.; Xu, B.; Wu, P.; Li, Q. Stability of the Al/TiB2 interface and doping effects of Mg/Si. Appl. Surf. Sci. 2017, 425, 639–645. [Google Scholar] [CrossRef]
- Bai, P.; Wang, Q.; Zhao, Z.; Du, W.; Liang, M.; Liao, H.; Li, Y.; Zhang, L.; Han, B.; Li, J. Interfacial stability of TiC/γ-Fe in TiC/316L stainless steel composites prepared by selective laser melting: First principles and experiment. Metals 2020, 10, 1225. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Liang, Y.; Zhao, R.; Jiang, Q. Fabrication of steel matrix composites locally reinforced with different ratios of TiC/TiB2 particulates using SHS reactions of Ni-Ti-B_4C and Ni-Ti-B_4C-C systems during casting. Mater. Sci. Eng. A 2007, 445, 398–404. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Segall, M.; Lindan, P.; Probert, M.; Pickard, C.; Hasnip, P.; Clark, S.; Payne, M. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Saib, S.; Bouarissa, N. Electronic properties of GaN at high-pressure from local density and generalized gradient approximations. Comp. Mater. Sci. 2006, 376, 13–617. [Google Scholar] [CrossRef]
- Pfrommer, B.; Côté, M.; Louie, S.; Cohen, M. Relaxation of Crystals with the Quasi-Newton Method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Li, Y.; Peng, J.; Sun, L.; Li, B.; Wang, Z.; Zhao, S. A comparable study of Fe//MCs (M = Ti, V) interfaces by first-principles method: The chemical bonding, work of adhesion and electronic structures. J. Phys. Chem. Solids 2020, 138, 109292. [Google Scholar] [CrossRef]
- Chung, S.; Ha, H.; Jung, W.; Byun, J. An ab Initio Study of the Energetics for Interfaces between Group V Transition Metal Carbides and bcc Iron. Model. Simul. Mater. Sci. 2006, 46, 1523–1531. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Han, Y.; Wang, X.; Liu, W.; Wu, J.; Cui, H. First-Principles Study of Adhesion Strength and Stability of the TiB2/TiC Interface in Composite Materials. Ceram. Int. 2018, 44, 1756–1763. [Google Scholar] [CrossRef]
- Otani, S.; Ishizawa, Y. Preparation of TiB2 single crystals by the floating zone method. J. Cryst. Growth 1994, 140, 451–453. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, P.; Zhou, Y.; Guo, J.; Ren, X.; Yang, Y.; Yang, Q. First-principles study on ferrite/TiC heterogeneous nucleation interface. J. Alloys Compd. 2013, 556, 160–166. [Google Scholar] [CrossRef]
- Shi, Z.; Liu, S.; Zhou, Y.; Yang, Q. First-principles calculation on the relationships of h-WC/γ-Fe interface. J. Phys. Chem. Solids 2018, 123, 11–18. [Google Scholar] [CrossRef]
- Xiong, H.; Zhang, H.; Dong, J. Adhesion strength and stability of TiB2/TiC interface in composite coatings by first principles calculation. Comp. Mater. Sci. 2017, 127, 244–250. [Google Scholar] [CrossRef]
- Wang, C.; Wang, C. Ni/Ni3Al interface: A density functional theory study. Appl. Surf. Sci. 2009, 255, 3669–3675. [Google Scholar] [CrossRef]
- Choe, W.; Miller, G.; Levin, E. Crystal Structure and Magnetism of Gd2MgGe2. J. Alloys Compd. 2001, 329, 121–130. [Google Scholar] [CrossRef]
- Wang, N.; Dong, L.; Gao, C.; Li, D. A study of structure, energy and electronic properties of TiB2/c-BN interface by first principles calculations. Opt. Mater. 2014, 36, 1459–1462. [Google Scholar] [CrossRef]
- Xiong, H.; Zhang, H.; Zhang, H.; Zhou, Y. Effects of alloying elements X (X=Zr, V, Cr, Mn, Mo, W, Nb, Y) on ferrite/TiC heterogeneous nucleation interface: First-principles study. J. Iron Steel Res. Int. 2017, 24, 328–334. [Google Scholar] [CrossRef]
- Jiang, Q.; Lu, H. Size dependent interface energy and its applications. Surf. Sci. Rep. 2008, 63, 427–464. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Method | A (Å) | c (Å) | V0 (Å3) | B (GPa) | ΔrH (eV/atom) |
|---|---|---|---|---|---|---|
| γ-Fe | GGAthis work | 3.445 | 3.445 | 40.85 | 306 | - |
| GGA [24] | 3.448 | 3.448 | 41.01 | 314.7 | - | |
| Exp [25] | 3.450 | 3.450 | 41.06 | - | - | |
| TiB2 | GGAthis work | 3.029 | 3.228 | 25.655 | 231.53 | −1.10 |
| GGA [26] | 3.033 | 3.231 | 25.73 | 260.5 | −1.04 | |
| Exp [27] | 3.03 | 3.229 | 25.67 | - | - |
| Layer (N) | Surface Energy (J/m2) |
|---|---|
| γ-Fe(111) | |
| 5 | 2.69 |
| 7 | 2.56 |
| 9 | 2.55 |
| 11 | 2.55 |
| Surface | Termination | Interlayer | Thickness (N) | ||||
|---|---|---|---|---|---|---|---|
| 3 | 5 | 7 | 9 | 11 | |||
| TiB2 | Ti | Δ1–2 | −5.48 | −6.04 | −5.56 | −5.27 | −6.49 |
| Δ2–3 | - | −1.62 | −3.33 | −1.46 | −2.27 | ||
| Δ3–4 | - | - | −0.89 | −2.84 | −2.27 | ||
| Δ4–5 | - | - | - | −1.05 | −4.54 | ||
| Δ5–6 | - | - | - | −0.34 | |||
| B | Δ1–2 | −3.45 | −6.45 | −4.34 | −4.26 | −4.99 | |
| Δ2–3 | - | −2.60 | −1.18 | −1.50 | −1.30 | ||
| Δ3–4 | - | - | −0.39 | −1.99 | −1.91 | ||
| Δ4–5 | - | - | - | −0.38 | −1.70 | ||
| Δ5–6 | - | - | - | - | −0.84 | ||
| Termination | Stacking Sequences | After Relaxation | |
|---|---|---|---|
| d0 (Å) | Wad (J/m2) | ||
| B-terminated | top 1 | 1.99 | 2.6 |
| top 2 | 1.24 | 4.16 | |
| center | 2 | 2.62 | |
| Ti-terminated | top 1 | 2.14 | 1.58 |
| top 2 | 2.11 | 3.2 | |
| center | 2.13 | 1.76 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Bai, P.; Zhao, Z. First Principle Study of TiB2 (0001)/γ-Fe (111) Interfacial Strength and Heterogeneous Nucleation. Materials 2021, 14, 1573. https://doi.org/10.3390/ma14061573
Wang Q, Bai P, Zhao Z. First Principle Study of TiB2 (0001)/γ-Fe (111) Interfacial Strength and Heterogeneous Nucleation. Materials. 2021; 14(6):1573. https://doi.org/10.3390/ma14061573
Chicago/Turabian StyleWang, Qin, Peikang Bai, and Zhanyong Zhao. 2021. "First Principle Study of TiB2 (0001)/γ-Fe (111) Interfacial Strength and Heterogeneous Nucleation" Materials 14, no. 6: 1573. https://doi.org/10.3390/ma14061573