
Structure Prediction and Mechanical Properties of Silicon Hexaboride on Ab Initio Level

 and
and
Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
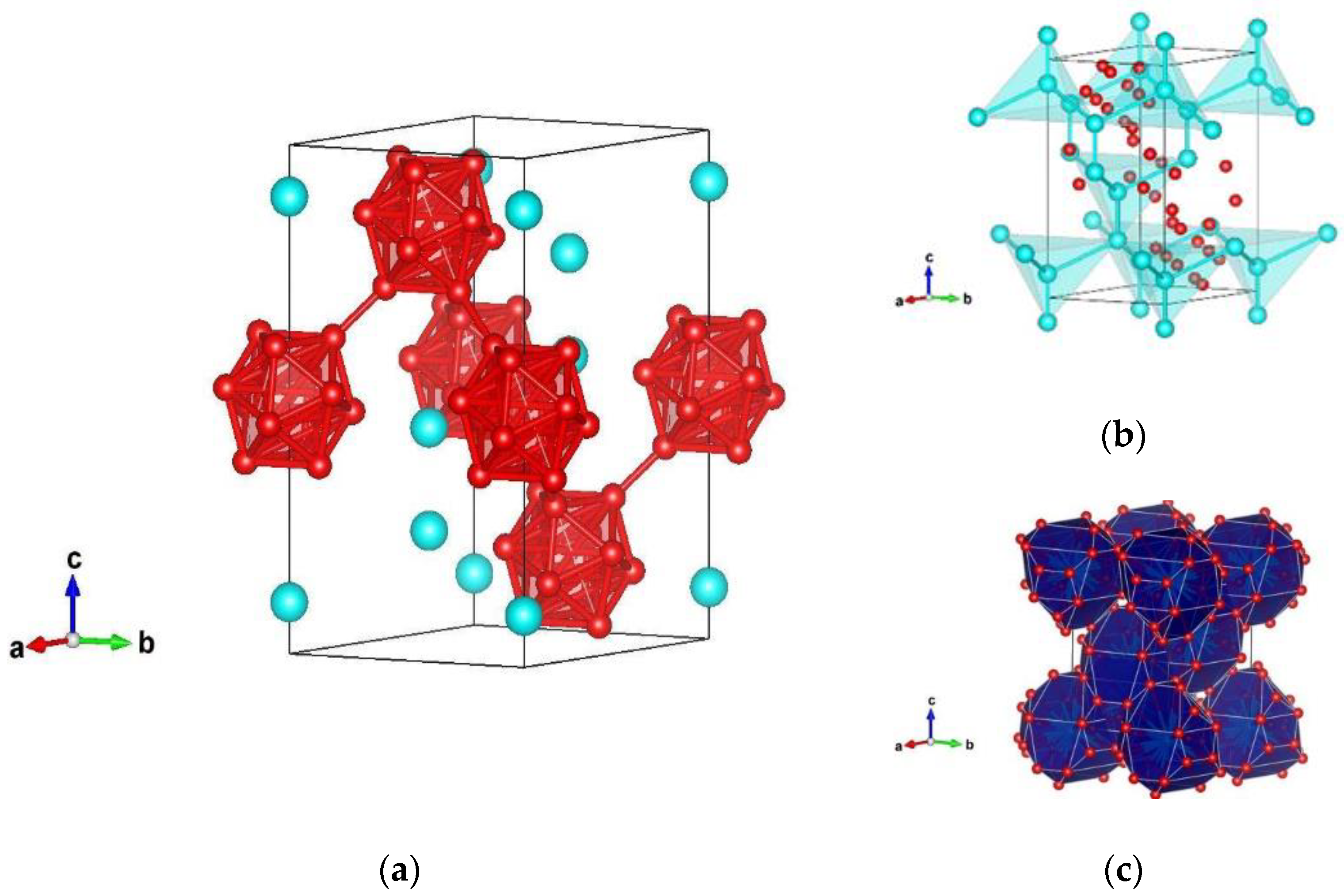
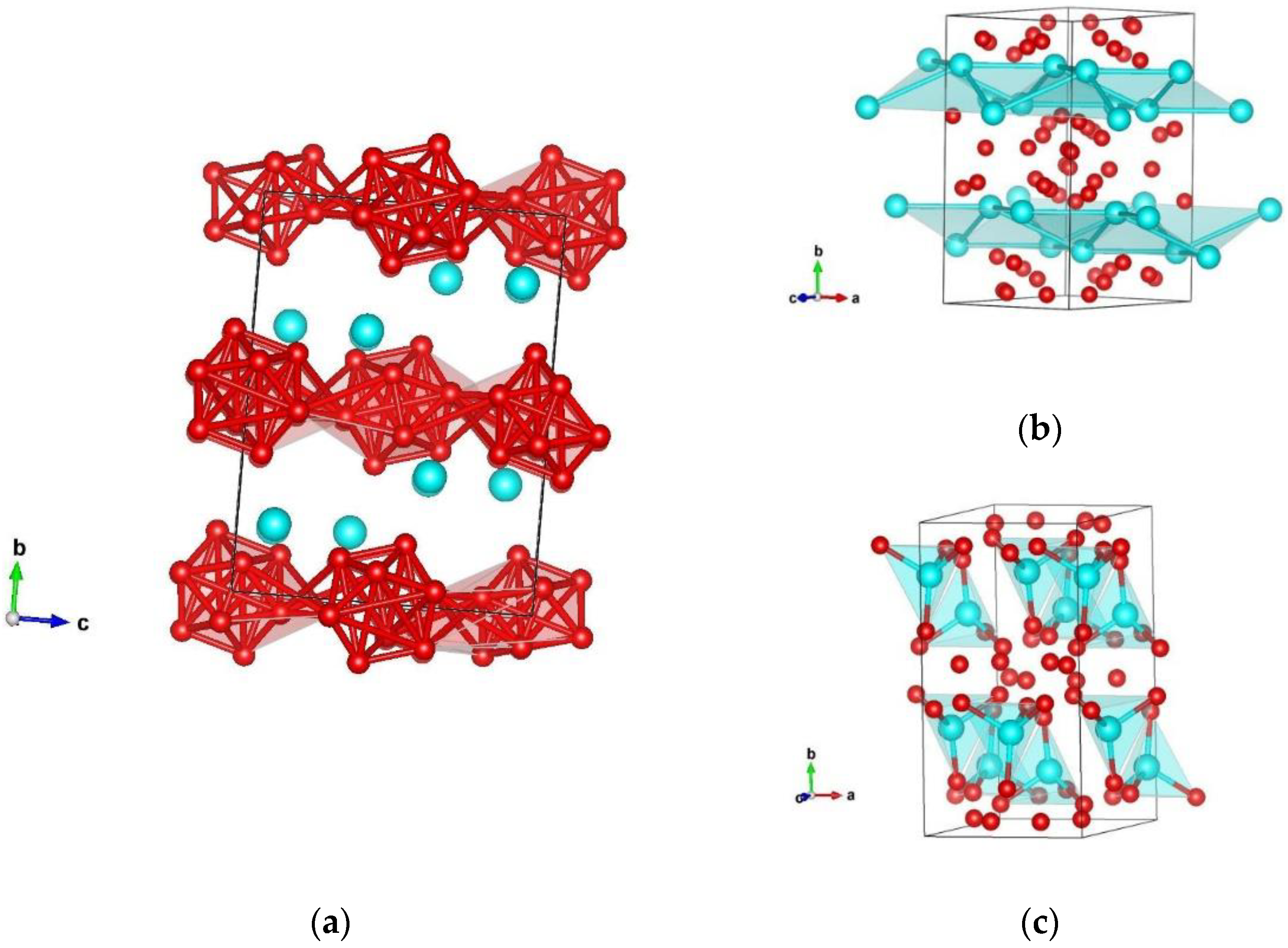
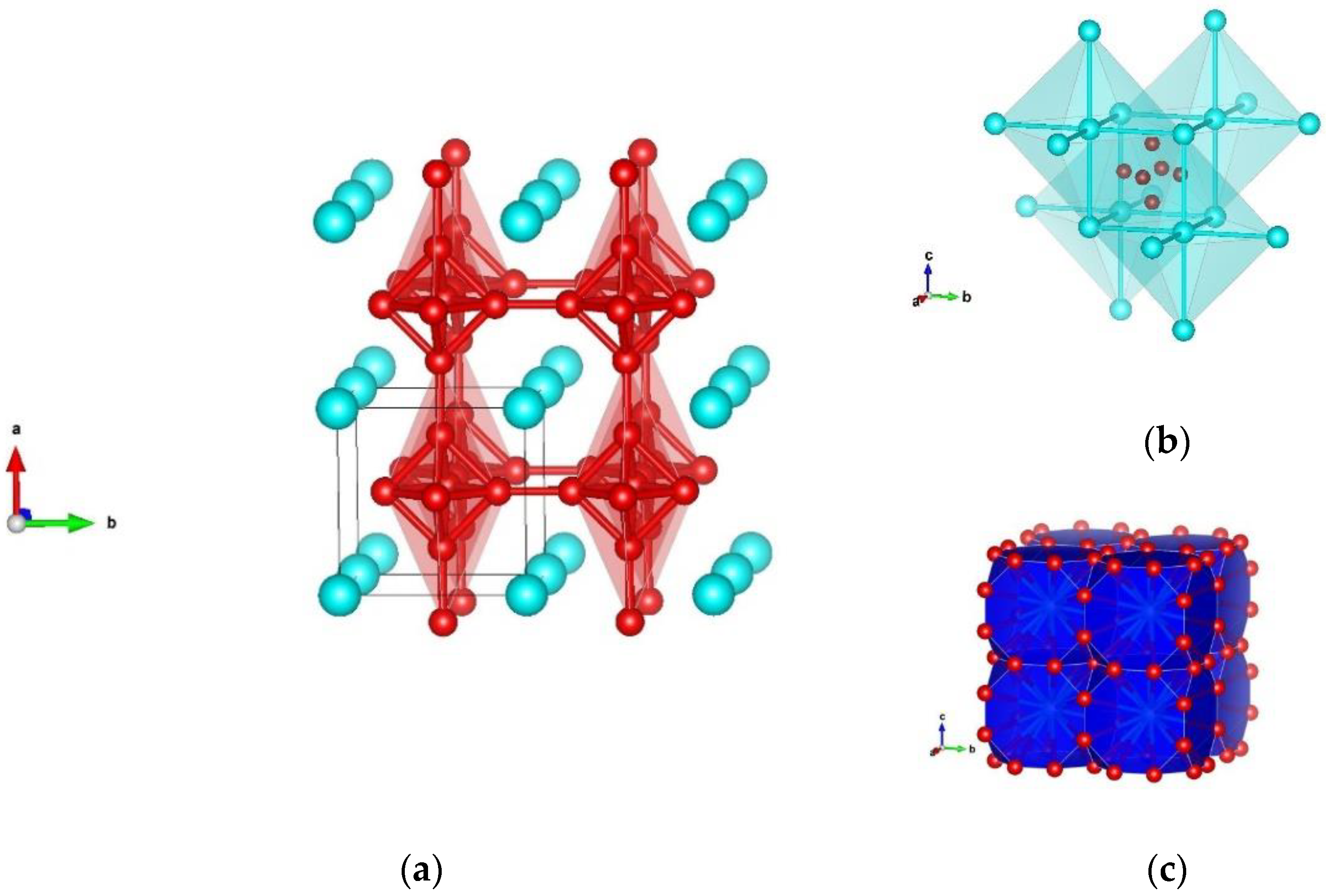
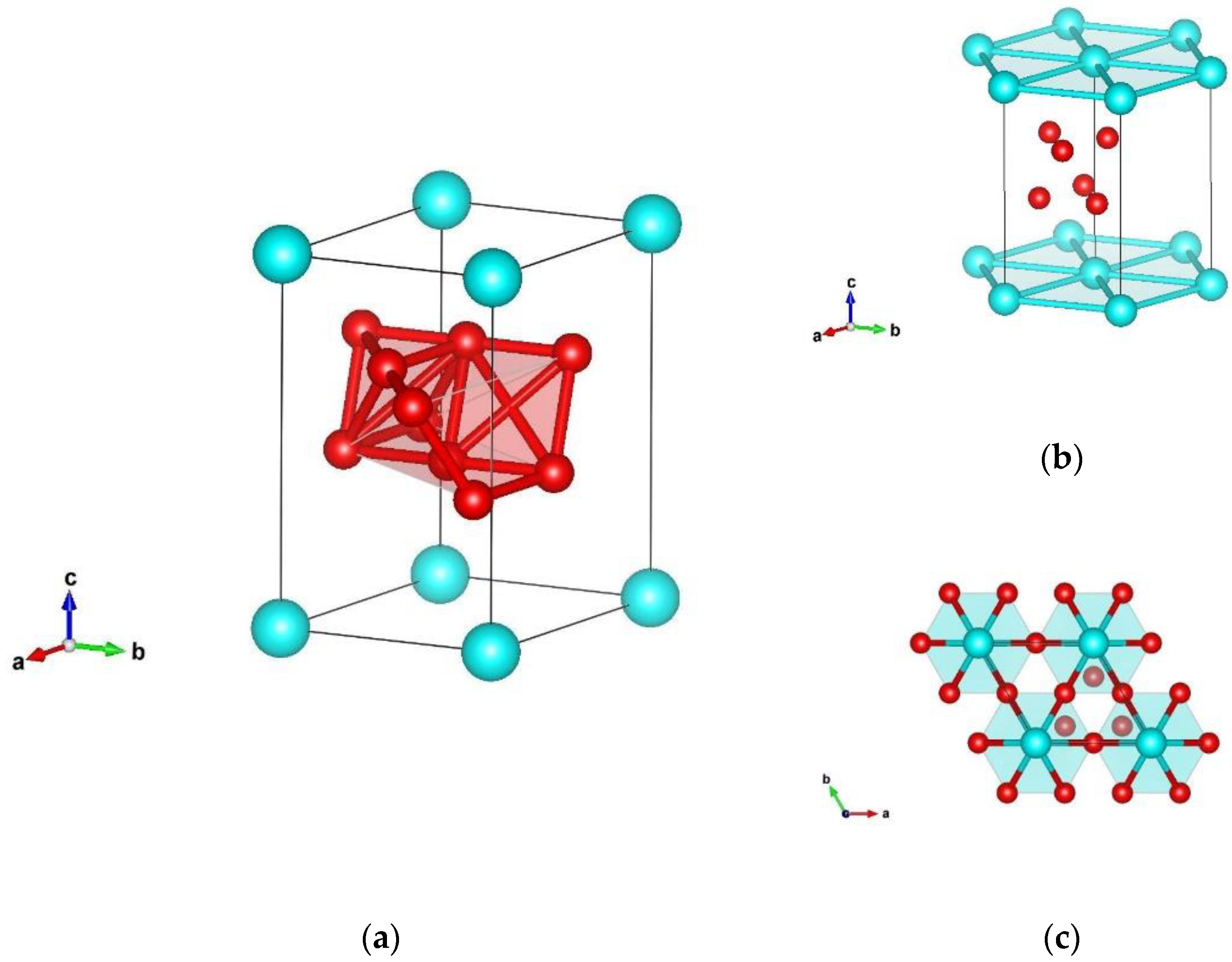
3.1. Structure Prediction of Silicon Hexaboride
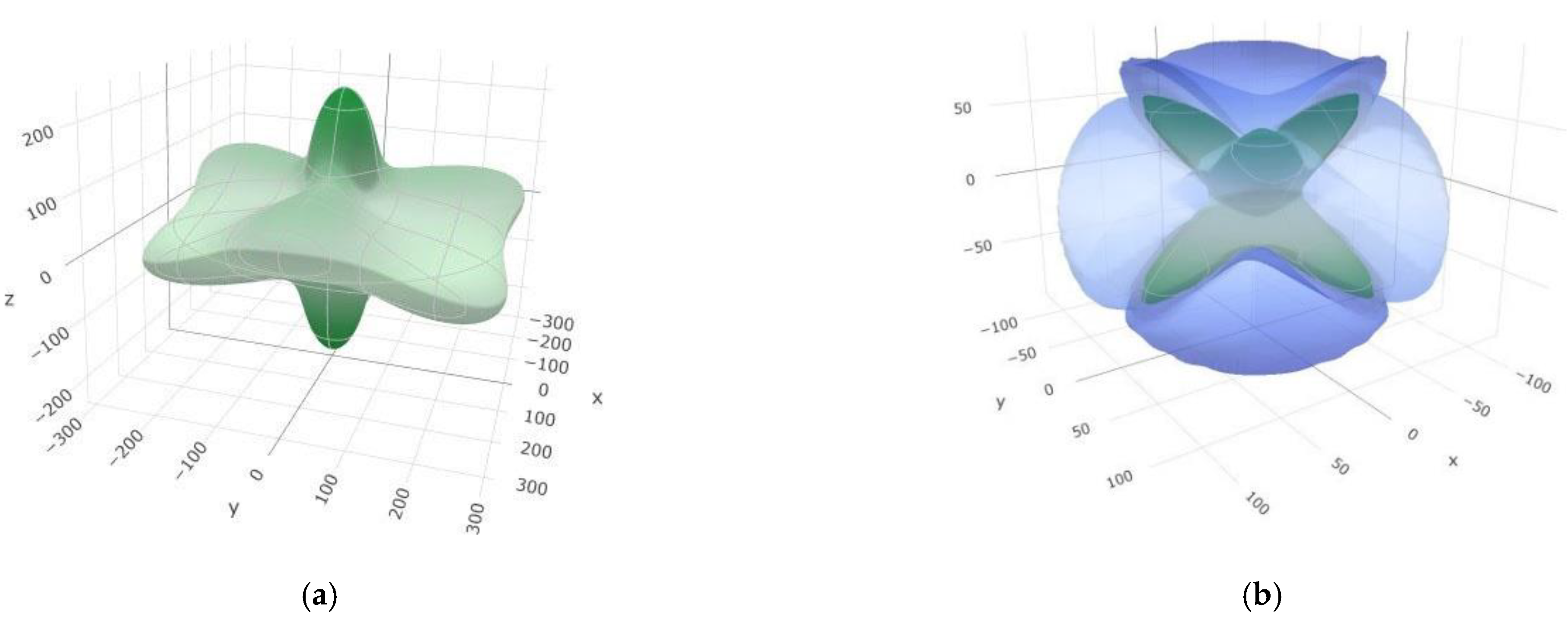
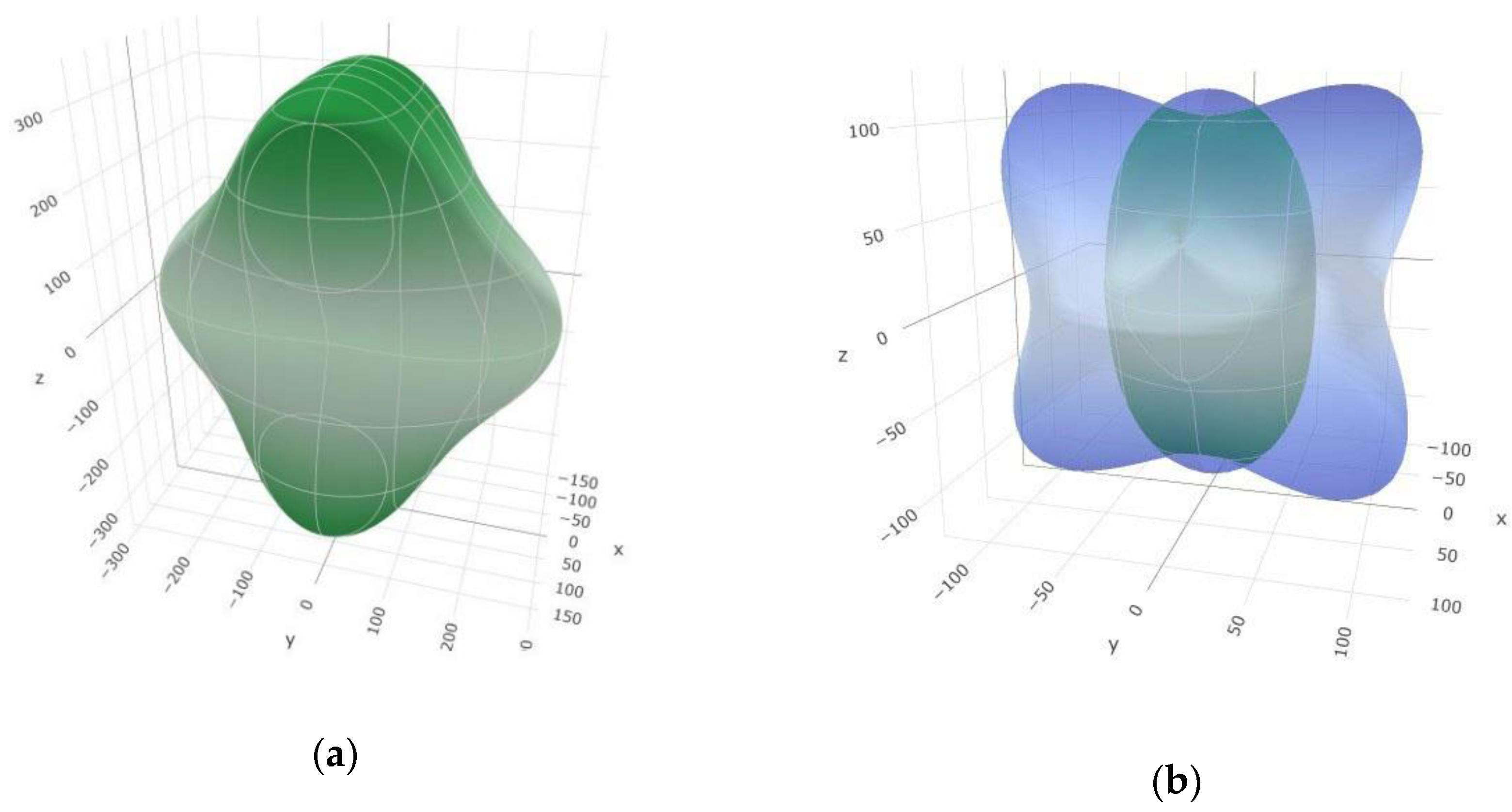
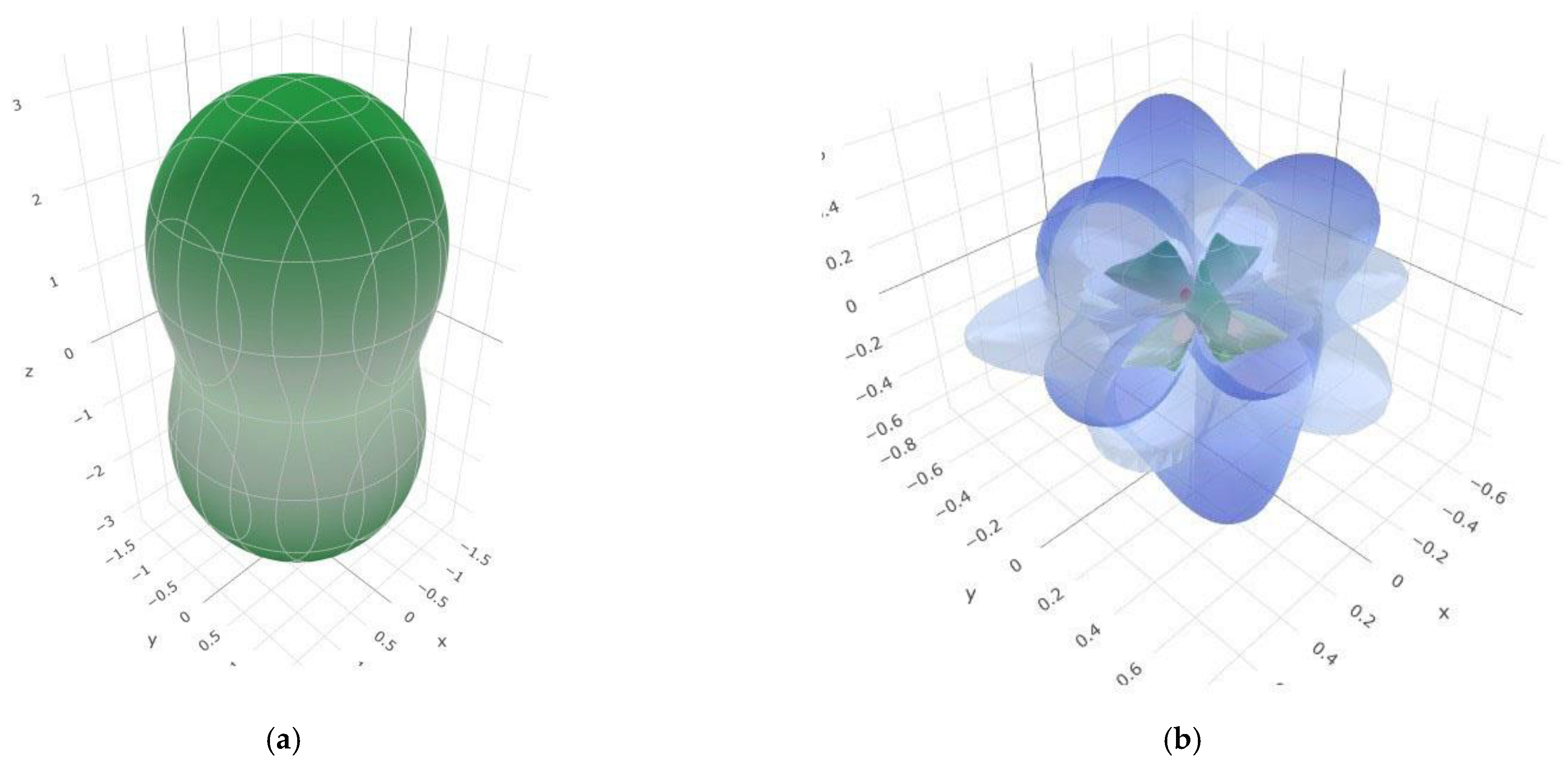
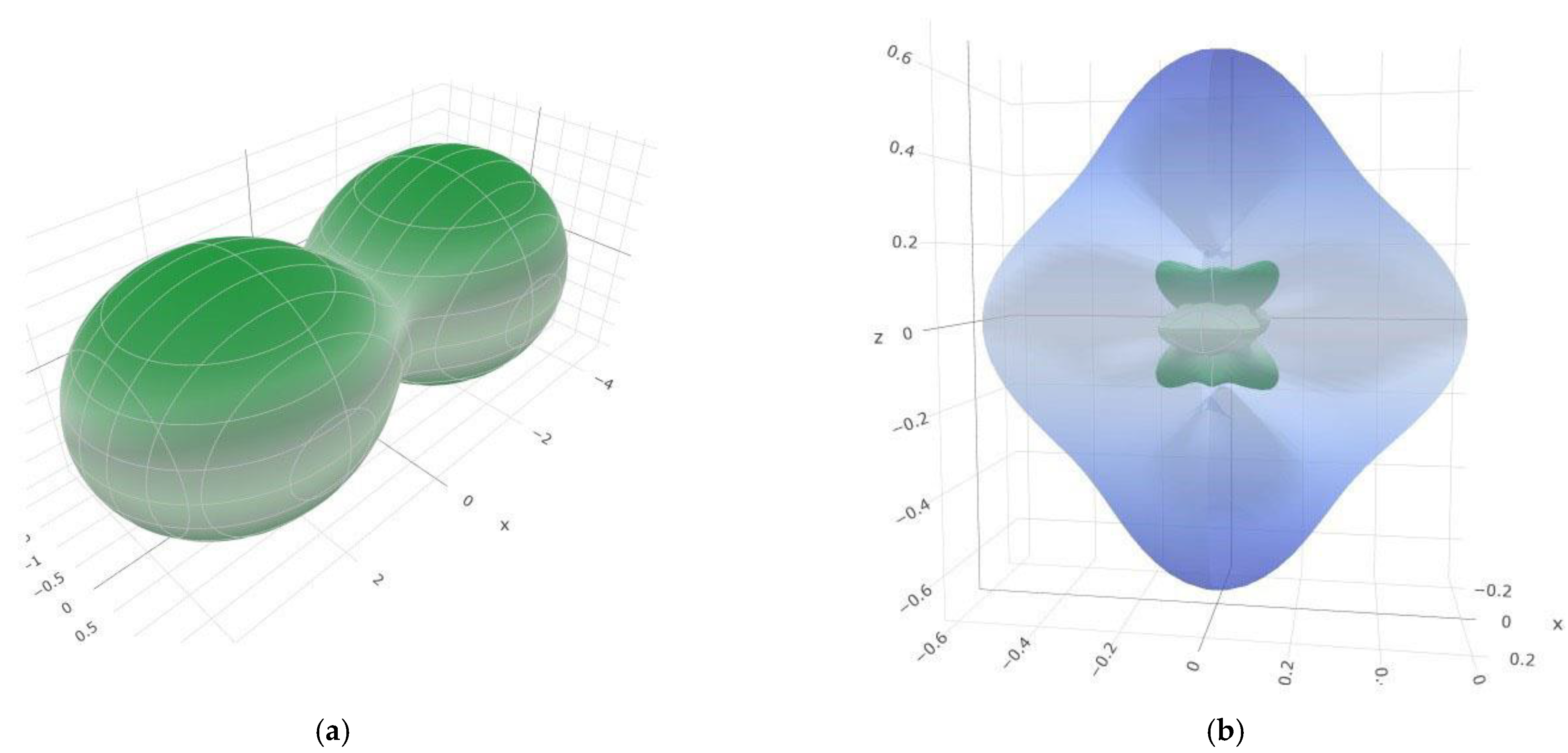
3.2. Elastic and Mechanical Properties of SiB6
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification and Structure Type | Space Group | Cell Parameters | Position of Atoms |
|---|---|---|---|
| α-SiB6 PB6-type | R-3mH (no. 166) | a = 6.160 c = 11.690 | Si 0.000000 0.000000 0.868964 B 0.149903 0.299806 0.528563 B −0.103858 −0.207715 0.618537 |
| β-SiB6 Cmca-B6Si | Cmce (no. 64) | a = 5.790 b = 11.053 c = 8.322 | Si 0.000000 0.824075 0.875433 B 0.736220 0.925098 0.973414 B 0.341461 0.038884 0.830830 B 0.000000 0.650086 0.938827 B 0.000000 0.396399 0.850014 |
| γ-SiB6 CaB6 type | Pm-3m (no. 221) | a = 4.109 | Si 0.000000 0.000000 0.000000 B 0.799534 0.500000 0.500000 |
| δ-SiB6 | P3m1 (no. 156) | a = 3.465 c = 6.059 | Si 0.000000 0.000000 0.000000 B 0.831926 0.663851 0.439282 B 0.999194 0.499597 0.712537 |
| Cij | GGA-PBE (GPa) | ||
|---|---|---|---|
| α-SiB6 | β-SiB6 | γ-SiB6 | |
| C11 | 80.39 | 187.47 | 362.06 |
| C12 | −90.35 | 83.66 | 38.62 |
| C13 | 207.40 | 100.87 | 38.62 |
| C15 | 10.28 | - | - |
| C21 | - | 86.67 | - |
| C22 | - | 322.35 | - |
| C23 | - | 53.39 | - |
| C31 | 323.26 | 97.85 | - |
| C32 | - | 54.68 | - |
| C33 | 132.77 | 379.45 | - |
| C44 | 39.99 | 96.55 | −8.70 |
| C46 | - | - | - |
| C55 | - | 118.88 | - |
| C66 | 85.37 | 64.39 | - |


Appendix B
References
- Cline, C.F. An investigation of the compound silicon boride (SiB6). J. Electrochem. Soc. 1959, 106, 322. [Google Scholar] [CrossRef]
- Takashima, N.; Azuma, Y.; Matsushita, J.-I. High-Temperature Thermoelectric Properties of Silicon Boride Ceramics as a Smart Material. MRS Proc. 2011, 604, 233. [Google Scholar] [CrossRef]
- Tanaka, S.; Fukushima, N.; Matsushita, J.-I.; Akatsu, T.; Niihara, K.; Yasuda, E. Mechanical properties of SiB6 addition of carbon sintered body. In Proceedings of the Smart Materials, Newport Beach, CA, USA, 4–8 March 2001; pp. 346–354. [Google Scholar]
- Zaitsev, A.; Kodentsov, A. Thermodynamic properties and phase equilibria in the Si-B system. J. Phase Equilibria 2001, 22, 126–135. [Google Scholar] [CrossRef]
- Olesinski, R.; Abbaschian, G. The B−Si (boron-silicon) system. Bull. Alloy Phase Diagr. 1984, 5, 478–484. [Google Scholar] [CrossRef]
- Imam, M.A.; Young, J.S.; Reddy, R.G. Determination of Thermodynamic Properties of Si-B Alloys. Metall. Mater. Trans. B 2019, 50, 981–990. [Google Scholar] [CrossRef]
- Moissan, H.; Stock, A. Preparation and properties of two silicon borides: SiB3 and SiB6. C. R. Acad. Sci. 1900, 131, 139–143. [Google Scholar]
- Zhuravlev, N. X-ray determination of the structure of SiB. Kristallografiya 1956, 1, 666–668. [Google Scholar]
- Zhang, B.; Wu, L.; Li, Z. Predicted structural evolution and detailed insight into configuration correlation, mechanical properties of silicon–boron binary compounds. RSC Adv. 2017, 7, 16109–16118. [Google Scholar] [CrossRef] [Green Version]
- Vlasse, M.; Slack, G.A.; Garbauskas, M.; Kasper, J.S.; Viala, J.C. The crystal structure of SiB6. J. Solid State Chem. 1986, 63, 31–45. [Google Scholar] [CrossRef]
- Durandurdu, M. Amorphous silicon hexaboride: A first-principles study. Philos. Mag. 2018, 98, 2723–2733. [Google Scholar] [CrossRef]
- Mirzayev, M.; Jabarov, S.; Asgerov, E.; Mehdiyeva, R.; Thabethe, T.T.; Biira, S.; Tiep, N. Crystal structure changes and weight kinetics of silicon-hexaboride under gamma irradiation dose. Results Phys. 2018, 10, 541–545. [Google Scholar] [CrossRef]
- Mirzayev, M.; Jabarov, S.; Asgerov, E.; Mehdiyeva, R.; Thabethe, T.T.; Biira, S.; Tiep, N. X-ray diffraction and thermodynamics kinetics of SiB6 under gamma irradiation dose. Silicon 2019, 11, 2499–2504. [Google Scholar] [CrossRef] [Green Version]
- Mirzayev, M.; Mammadov, K.F.; Skuratov, V.; Demir, E.; Jabarov, S.; Ismayilova, N.; Biira, S.; Abdurakhimov, B.; Popov, E. Oxidation kinetics and thermophysical properties of gamma irradiated silicon hexaboride. J. Alloys Compd. 2019, 801, 151–157. [Google Scholar] [CrossRef]
- Durandurdu, M. Amorphous silicon hexaboride at high pressure. Philos. Mag. 2020, 100, 1818–1833. [Google Scholar] [CrossRef]
- Imai, Y.; Mukaida, M.; Ueda, M.; Watanabe, A. Band-calculation of the electronic densities of states and the total energies of boron–silicon system. J. Alloys Compd. 2002, 347, 244–251. [Google Scholar] [CrossRef]
- Yuan, Z.; Xiong, M.; Yu, D. A novel metallic silicon hexaboride, Cmca-B6Si. Phys. Lett. A 2020, 384, 126075. [Google Scholar] [CrossRef]
- Imam, M.A.; Reddy, R.G. A Review of Boron-Rich Silicon Borides Basedon Thermodynamic Stability and Transport Properties of High-Temperature Thermoelectric Materials. High Temp. Mater. Process. 2019, 38, 411–424. [Google Scholar] [CrossRef]
- Lee, D.; Won, J.; Kim, K.; Matsushita, J.; Shim, K. Microstructural evolution of La-doped SiB6 high-temperature thermoelectric material during a Spark Plasma Sintering. MRS Online Proc. Libr. (OPL) 2001, 691. [Google Scholar] [CrossRef]
- Mukaida, M.; Tsunoda, T.; Imai, Y. Preparation of B-Si films by chemical vapor deposition. In Proceedings of the Eighteenth International Conference on Thermoelectrics, Baltimore MD, USA, 29 August–2 September 1999; pp. 667–670. [Google Scholar]
- Shuang, S.; Yang, F.; Li, Z.; Li, J.; Meng, X. Synthesis and Infrared Performance of SiB6 Powder through “Chemical Oven” Self-Propagating Combustion. Adv. Mater. Sci. Eng. 2021, 2021, 9991967. [Google Scholar] [CrossRef]
- Hu, P.; Zhao, P.-P.; Jin, Y. Thermophysical properties of pentaerythritol/nano-SiB 6 composites for thermal storage. In Proceedings of the 14th IEEE International Conference on Nanotechnology, Toronto, ON, Canada, 18–21 August 2014; pp. 732–735. [Google Scholar]
- Wang, L.; Wang, W.; Fu, Q. The improvement of the self-healing ability of MoSi2 coatings at 900–1200 °C by introducing SiB6. J. Eur. Ceram. Soc. 2020, 40, 2896–2906. [Google Scholar] [CrossRef]
- Matsushita, J.; Komarneni, S. High temperature oxidation of silicon hexaboride ceramics. Mater. Res. Bull. 2001, 36, 1083–1089. [Google Scholar] [CrossRef]
- Kim, T.K.; Moon, J.; VanSaders, B.; Chun, D.; Gardner, C.J.; Jung, J.-Y.; Wang, G.; Chen, R.; Liu, Z.; Qiao, Y. Si boride-coated Si nanoparticles with improved thermal oxidation resistance. Nano Energy 2014, 9, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Bergerhoff, G.; Brown, I.; Allen, F. Crystallographic databases. Int. Union Crystallogr. Chester 1987, 360, 77–95. [Google Scholar]
- Zagorac, D.; Müller, H.; Ruehl, S.; Zagorac, J.; Rehme, S. Recent developments in the Inorganic Crystal Structure Database: Theoretical crystal structure data and related features. J. Appl. Crystallogr. 2019, 52, 918–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagorac, J.; Schön, J.C.; Matović, B.; Škundrić, T.; Zagorac, D. Predicting Feasible Modifications of Ce2ON2 Using a Combination of Global Optimization and Data Mining. J. Phase Equilibria Diffus. 2020, 41, 538–549. [Google Scholar] [CrossRef]
- Zagorac, J.; Zagorac, D.; Rosić, M.; Schön, J.C.; Matović, B. Structure prediction of aluminum nitride combining data mining and quantum mechanics. CrystEngComm 2017, 19, 5259–5268. [Google Scholar] [CrossRef]
- Cvijović-Alagić, I.; Rakin, M.; Laketić, S.; Zagorac, D. Microstructural study of Ti-45Nb alloy before and after HPT processing using experimental and ab initio data mining approach. Mater. Charact. 2020, 169, 110635. [Google Scholar] [CrossRef]
- Škundrić, T.; Zagorac, D.; Schön, J.C.; Pejić, M.; Matović, B. Crystal Structure Prediction of the Novel Cr2SiN4 Compound via Global Optimization, Data Mining, and the PCAE Method. Crystals 2021, 11, 891. [Google Scholar] [CrossRef]
- Sokol, A.A.; Catlow, C.R.A.; Miskufova, M.; Shevlin, S.A.; Al-Sunaidi, A.A.; Walsh, A.; Woodley, S.M. On the problem of cluster structure diversity and the value of data mining. Phys. Chem. Chem. Phys. 2010, 12, 8438–8445. [Google Scholar] [CrossRef]
- Ceder, G.; Morgan, D.; Fischer, C.; Tibbetts, K.; Curtarolo, S. Data-mining-driven quantum mechanics for the prediction of structure. MRS Bull. 2006, 31, 981–985. [Google Scholar] [CrossRef] [Green Version]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S. Quantum-mechanical condensed matter simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Dovesi, R.; Orlando, R.; Civalleri, B.; Roetti, C.; Saunders, V.R.; Zicovich-Wilson, C.M. CRYSTAL: A computational tool for the ab initio study of the electronic properties of crystals. Z. Für Krist.-Cryst. Mater. 2005, 220, 571–573. [Google Scholar] [CrossRef]
- Dovesi, R.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.; D’arco, P.; Noël, Y.; Rérat, M.; Carbonniere, P. The CRYSTAL code, 1976–2020 and beyond, a long story. J. Chem. Phys. 2020, 152, 204111. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048. [Google Scholar] [CrossRef] [Green Version]
- Pascale, F.; Zicovich-Wilson, C.M.; Orlando, R.; Roetti, C.; Ugliengo, P.; Dovesi, R. Vibration frequencies of Mg3Al2Si3O12 pyrope. An ab initio study with the CRYSTAL code. J. Phys. Chem. B 2005, 109, 6146–6152. [Google Scholar] [CrossRef] [PubMed]
- Noel, Y.; Catti, M.; D’Arco, P.; Dovesi, R. The vibrational frequencies of forsterite Mg 2 SiO 4: An all-electron ab initio study with the CRYSTAL code. Phys. Chem. Miner. 2006, 33, 383–393. [Google Scholar] [CrossRef]
- Doll, K.; Schön, J.; Jansen, M. Structure prediction based on ab initio simulated annealing for boron nitride. Phys. Rev. B 2008, 78, 144110. [Google Scholar] [CrossRef] [Green Version]
- Zagorac, J.; Matovic, B.; Pejic, M.; Milutinovic, K.; Zagorac, D. Crystal structure and properties of theoretically predicted AlB12. J. Innov. Mater. Extrem. Cond. 2020, 1, 28–36. [Google Scholar]
- Jovanović, D.; Zagorac, J.; Matović, B.; Zarubica, A.; Zagorac, D. Structural, electronic and mechanical properties of superhard B4C from first principles. J. Innov. Mater. Extrem. Cond. 2020, 1, 19–27. [Google Scholar]
- Zagorac, D.; Zagorac, J.; Doll, K.; Čebela, M.; Matović, B. Extreme pressure conditions of bas based materials: Detailed study of structural changes, band gap engineering, elastic constants and mechanical properties. Process. Appl. Ceram. 2019, 13, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Jovanović, D.; Zagorac, D.; Schön, J.C.; Milovanović, B.; Zagorac, J. A new theoretical model for hexagonal ice, Ih(d), from first principles investigations. Z. Nat. B 2020, 75, 125–128. [Google Scholar] [CrossRef] [Green Version]
- Zagorac, J.; Jovanović, D.; Volkov-Husović, T.; Matović, B.; Zagorac, D. Structure prediction, high pressure effect and properties investigation of superhard B6O. Model. Simul. Mater. Sci. Eng. 2020, 28, 035004. [Google Scholar] [CrossRef]
- Matović, B.; Luković, J.; Zagorac, D.; Ivanova, O.S.; Baranchikov, A.E.; Shekunova, T.O.; Yorov, K.E.; Gajtko, O.M.; Yang, L.; Rumyantseva, M.N.; et al. Crystalline WO3 nanoparticles for NO2 sensing. Process. Appl. Ceram. 2020, 14, 282–292. [Google Scholar] [CrossRef]
- Matović, B.; Zagorac, D.; Cvijović-Alagić, I.; Zagorac, J.; Butulija, S.; Erčić, J.; Hanzel, O.; Sedlák, R.; Lisnichuk, M.; Tatarko, P. Fabrication and characterization of high entropy pyrochlore ceramics. Boletín Soc. Española Cerámica Vidr. 2021, in press. [Google Scholar] [CrossRef]
- Perger, W.; Criswell, J.; Civalleri, B.; Dovesi, R. Ab-initio calculation of elastic constants of crystalline systems with the CRYSTAL code. Comput. Phys. Commun. 2009, 180, 1753–1759. [Google Scholar] [CrossRef]
- Erba, A.; Mahmoud, A.; Orlando, R.; Dovesi, R. Elastic properties of six silicate garnet end members from accurate ab initio simulations. Phys. Chem. Miner. 2014, 41, 151–160. [Google Scholar] [CrossRef]
- Gaillac, R.; Pullumbi, P.; Coudert, F.-X. ELATE: An open-source online application for analysis and visualization of elastic tensors. J. Phys. Condens. Matter 2016, 28, 275201. [Google Scholar] [CrossRef]
- Hundt, R.; SchoÈn, J.C.; Hannemann, A.; Jansen, M. Determination of symmetries and idealized cell parameters for simulated structures. J. Appl. Crystallogr. 1999, 32, 413–416. [Google Scholar] [CrossRef] [Green Version]
- Hannemann, A.; Hundt, R.; Schön, J.; Jansen, M. A new algorithm for space-group determination. J. Appl. Crystallogr. 1998, 31, 922–928. [Google Scholar] [CrossRef]
- Hundt, R. KPLOT: A Program for Plotting and Analysing Crystal Structures; Technicum Scientific Publishing: Stuttgart, Germany, 2016. [Google Scholar]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Zagorac, D.; Zagorac, J.; Schön, J.C.; Stojanović, N.; Matović, B. ZnO/ZnS (hetero) structures: Ab initio investigations of polytypic behavior of mixed ZnO and ZnS compounds. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2018, 74, 628–642. [Google Scholar] [CrossRef]
- Magnusson, B.; Brosset, C. The Crystal Structure of B2.89Si. Acta Chem. Scand. 1962, 16, 449–455. [Google Scholar] [CrossRef]
- Matkovich, V.I. Interstitial Compounds of Boron. J. Am. Chem. Soc. 1961, 83, 1804–1806. [Google Scholar] [CrossRef]
- Zagorac, D.; Doll, K.; Schön, J.C.; Jansen, M. Ab initio structure prediction for lead sulfide at standard and elevated pressures. Phys. Rev. B 2011, 84, 045206. [Google Scholar] [CrossRef]
- Jemmis, E.D.; Prasad, D.L.V.K. Icosahedral B12, macropolyhedral boranes, β-rhombohedral boron and boron-rich solids. J. Solid State Chem. 2006, 179, 2768–2774. [Google Scholar] [CrossRef]
- Dai, J.; Tian, Z. Large thermal conductivity of boron suboxides despite complex structures. Appl. Phys. Lett. 2021, 118, 041901. [Google Scholar] [CrossRef]
- Luo, R.-B.; Zeng, W.; Tang, B.; Zhong, M.; Liu, Q.-J. Electronic structures, effective masses and optical properties of B6Ch (Ch=O, S, Se, Te) based on DFT study. Solid State Commun. 2021, 336, 114423. [Google Scholar] [CrossRef]
- Han, H. Density-functional theory study of the effect of pressure on the elastic properties of CaB6. Chin. Phys. B 2013, 22, 077101. [Google Scholar] [CrossRef]
- Moradkhani, A.; Baharvandi, H. Determining the fracture resistance of B4C-NanoSiB6 nanocomposite by Vickers indentation method and exploring its mechanical properties. Int. J. Refract. Met. Hard Mater. 2017, 68, 159–165. [Google Scholar] [CrossRef]
- Murakami, T.; Inui, H. Friction and wear properties of spark-plasma-sintered α-AlB12 and SiB6 powder compacts in water. Tribol. Int. 2015, 92, 446–453. [Google Scholar] [CrossRef]
- Murakami, T.; Korenaga, A.; Ohana, T.; Inui, H. Tribological properties of aluminum and silicon borides at high temperatures. In Materials Science Forum; Trans Tech Publications Ltd.: Freienbach, Switzerland, 2018; pp. 1984–1989. [Google Scholar]
- Novikov, N.; Voronkin, M.; Zaika, N. Deposition of cBN films by ion sputtering of SiB6 and AlB12 boron-base materials. Diam. Relat. Mater. 1998, 7, 1693–1697. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef] [Green Version]
- Zagorac, J.; Zagorac, D.; Jovanović, D.; Luković, J.; Matović, B. Ab initio investigations of structural, electronic and mechanical properties of aluminum nitride at standard and elevated pressures. J. Phys. Chem. Solids 2018, 122, 94–103. [Google Scholar] [CrossRef]
- Ashby, M.F.; Shercliff, H.; Cebon, D. Materials: Engineering, Science, Processing and Design; Elsevier Science: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Zagorac, D.; Zagorac, J.; Djukic, M.; Jordanov, D.; Matović, B. Theoretical study of AlN mechanical behaviour under high pressure regime. Theor. Appl. Fract. Mech. 2019, 103, 102289. [Google Scholar] [CrossRef]
- Karacaoğlan, A.Ö.; Durandurdu, M. A first principles study of amorphous and crystalline silicon tetraboride. Mater. Chem. Phys. 2021, 258, 123928. [Google Scholar] [CrossRef]
- Pugh, S. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Vaitheeswaran, G.; Kanchana, V.; Kumar, R.S.; Cornelius, A.; Nicol, M.; Svane, A.; Delin, A.; Johansson, B. High-pressure structural, elastic, and electronic properties of the scintillator host material KMgF3. Phys. Rev. B 2007, 76, 014107. [Google Scholar] [CrossRef] [Green Version]






| Modifications | Total Energy | Relative Energy | ||
|---|---|---|---|---|
| GGA-PBE (Eh) | LDA-PZ (Eh) | GGA-PBE (Eh) | LDA-PZ (Eh) | |
| α-SiB6 | −438.2996 | −435.9498 | 0 | 0 |
| β-SiB6 | −438.2990 | −435.9432 | −0.0006 | −0.0066 |
| γ-SiB6 | −438.2009 | −435.8422 | −0.0987 | −0.1076 |
| δ-SiB6 | −438.1313 | −435.7806 | −0.1683 | −0.1692 |
| Modification and Structure Type | Space Group | Cell Parameters | Position of Atoms |
|---|---|---|---|
| α-SiB6 PB6-type | R-3mH (no. 166) | a = 6.164 c = 12.079 | Si 0.000000 0.000000 0.898652 |
| B 0.150405 0.300810 0.527650 | |||
| B –0.104630 −0.209260 0.618452 | |||
| β-SiB6 Cmca-B6Si | Cmce (no. 64) | a = 5.894 b = 11.184 c = 8.420 | Si 0.000000 0.825124 0.876686 |
| B 0.734869 0.925007 0.973036 | |||
| B 0.341293 0.039457 0.830441 | |||
| B 0.000000 0.650575 0.938946 | |||
| B 0.000000 0.396919 0.849121 | |||
| γ-SiB6 CaB6 type, SiB6-cubic | Pm-3m (no. 221) | a = 4.161 | Si 0.000000 0.000000 0.000000 |
| B 0.800175 0.500000 0.500000 | |||
| δ-SiB6 | P3m1 (no. 156) | a = 3.503 c = 6.407 | Si 0.000000 0.000000 0.000000 |
| B 0.831905 0.663809 0.465145 | |||
| B 0.999478 0.499739 0.726170 |
| Modification | Experiment/ Theory (Å) | GGA-PBE (Å) | LDA-PZ (Å) |
|---|---|---|---|
| α-SiB6 | n.a. | a = 6.164 c = 12.079 | a = 6.160 c = 11.690 |
| β-SiB6 | a = 5.8443 b = 11.0988 c = 8.3697 a | a = 5.894 b = 11.184 c = 8.420 | a = 5.790 b = 11.053 c = 8.322 |
| γ-SiB6 | a = 4.130 b a = 4.13 c | a = 4.161 | a = 4.109 |
| δ-SiB6 | n.a. | a = 3.503 c = 6.407 | a = 3.465 c = 6.059 |
| Cij (GPa) | LDA | ||
|---|---|---|---|
| α-SiB6 | β-SiB6 | γ-SiB6 | |
| C11 | 380.48 | 165.19 205 a | 404.76 402.6 b |
| C12 | 144.82 | 97.46 79 a | 32.55 19.31 b |
| C13 | 69.75 | 109.01 97 a | - |
| C15 | 28.99 | - | - |
| C21 | - | 101.32 | - |
| C22 | - | 352.39 338 a | - |
| C23 | - | 66.19 57 a | - |
| C31 | 69.94 | 112.28 | - |
| C32 | - | 68.81 | - |
| C33 | 249.68 | 409.08 397 a | - |
| C44 | 39.80 | 102.57 100 a | −11.62 −4.13 b |
| C46 | - | - | - |
| C55 | - | 117.45 123 a | - |
| C66 | 117.83 | 63.65 72 a | - |
| Mechanical Property | LDA | GGA | ||||
|---|---|---|---|---|---|---|
| α-SiB6 | β-SiB6 | γ-SiB6 | α-SiB6 | β-SiB6 | γ-SiB6 | |
| B | 169.21 | 154.59 | 156.63 | 153.43 | 147.22 | 146.44 |
| K | 71.55 | 92.09 | 23.63 | 47.22 | 93.88 | 22.21 |
| E | 188.12 | 230.49 | 67.49 | 128.48 | 232.26 | 63.42 |
| v | 0.32 | 0.25 | 0.43 | 0.36 | 0.24 | 0.43 |
| B/K | 2.36 | 1.68 | 6.63 | 3.25 | 1.57 | 6.59 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Škundrić, T.; Matović, B.; Zarubica, A.; Zagorac, J.; Tatarko, P.; Zagorac, D. Structure Prediction and Mechanical Properties of Silicon Hexaboride on Ab Initio Level. Materials 2021, 14, 7887. https://doi.org/10.3390/ma14247887
Škundrić T, Matović B, Zarubica A, Zagorac J, Tatarko P, Zagorac D. Structure Prediction and Mechanical Properties of Silicon Hexaboride on Ab Initio Level. Materials. 2021; 14(24):7887. https://doi.org/10.3390/ma14247887
Chicago/Turabian StyleŠkundrić, Tamara, Branko Matović, Aleksandra Zarubica, Jelena Zagorac, Peter Tatarko, and Dejan Zagorac. 2021. "Structure Prediction and Mechanical Properties of Silicon Hexaboride on Ab Initio Level" Materials 14, no. 24: 7887. https://doi.org/10.3390/ma14247887