
Ground State Properties of the Wide Band Gap Semiconductor Beryllium Sulfide (BeS)


Abstract
:1. Introduction
2. Computational Method and Related Details
3. Results
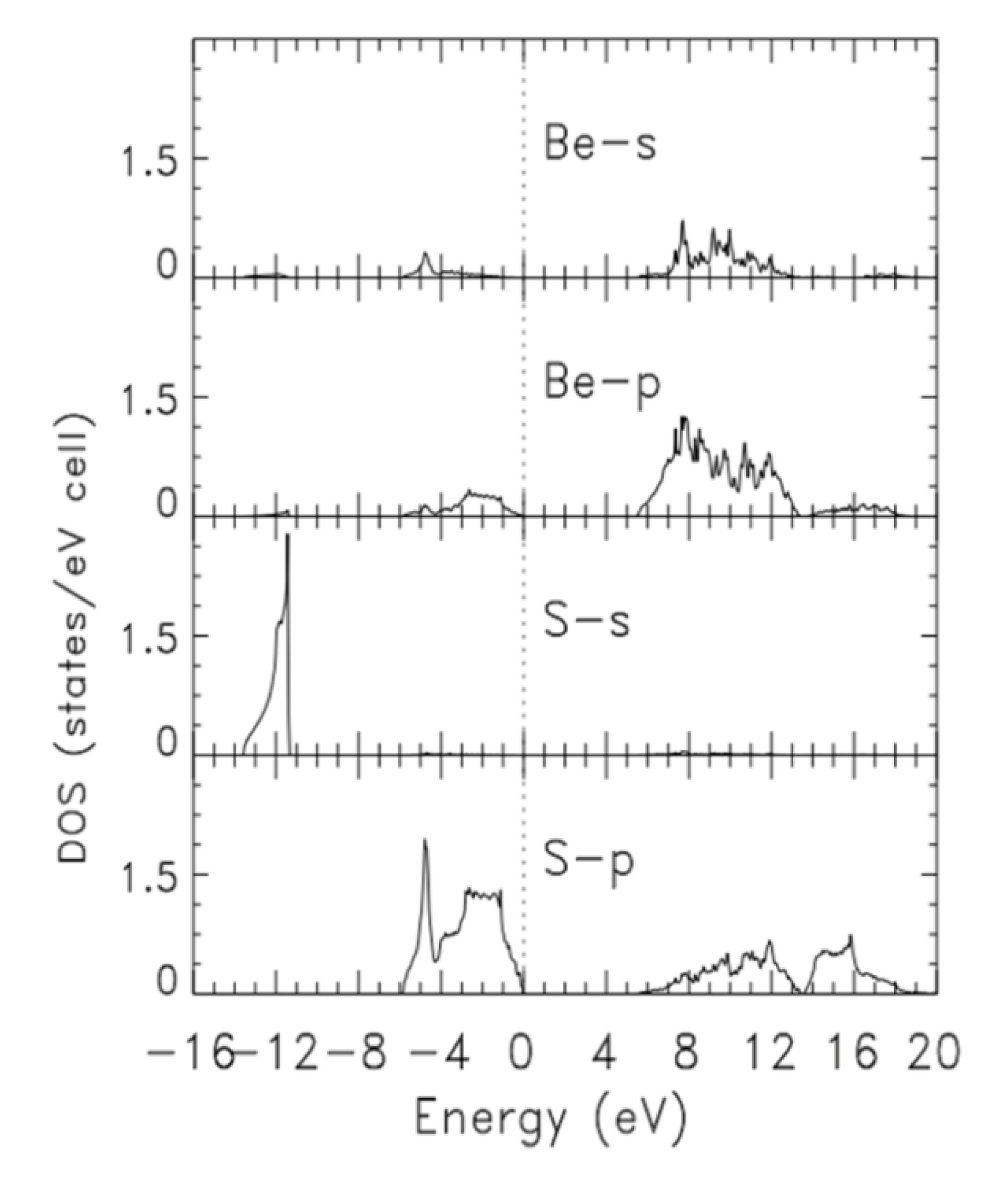
3.1. Electronic Properties
3.2. Transport Properties
3.3. Structural Properties
4. Discussion
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Landwehr, G. Molecular-beam epitaxy of beryllium-chalcogenide-based thin films and quantum-well structures. J. Appl. Phys. 1998, 792, 1–6. [Google Scholar]
- Ivanov, S.V.; Toropov, A.A.; Sorokin, S.V.; Shubina, T.V.; Il’inskaya, N.D.; Lebedev, A.V.; Sedova, I.V.; Kop’ev, P.S.; Alferov, Z.I.; Lugauer, H.J.; et al. Molecular beam epitaxy of alternating-strain ZnSe-based multilayer heterostructures for blue-green lasers. Semiconductors 1998, 32, 1137–1140. [Google Scholar] [CrossRef]
- Muñoz, A.; Rodríguez-Hernández, P.; Mujica, A. Ground-state properties and high-pressure phase of beryllium chalcogenides BeSe, BeTe, and BeS. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11861–11864. [Google Scholar] [CrossRef] [PubMed]
- Yim, W.; Dismukes, P.; Stofko, J.; Paff, J. Synthesis and some properties of BeTe, BeSe and BeS. J. Phys. Chem. Solids 1972, 33, 501–505. [Google Scholar] [CrossRef]
- Elias, B. Theoretical investigation of the structural, electronic, elastic, and optical properties of zinc- blende bes under high pressure. Theor. Investig. 2013, 2, 4193–4204. [Google Scholar]
- Gonzalez-Diaz, M.; Rodriguez-Hernandez, P.; Munoz, A. Elastic constants and electronic structure of beryllium chalcogenides BeS, BeSe, and BeTe from first-principles calculations. Phys. Rev. B 1996, 55, 14043. [Google Scholar] [CrossRef]
- Benosman, N.; Amrane, N.; Méçabih, S.; Aourag, H. Structural and electronic properties of bulk BeS. Phys. B Condens. Matter 2001, 304, 214–220. [Google Scholar] [CrossRef]
- Van Camp, P.E. Ground state properties and structural phase transformation of beryllium sulphide, solid state communication. J. Phys. Condens. Matter 1996, 98, 741–743. [Google Scholar]
- Van Vechten, J.A. Quantum dielectric theory of electronegativity in covalent systems. II. Ionization potentials and interband transition energies. Phys. Rev. 1969, 187, 1007–1020. [Google Scholar] [CrossRef]
- Skutel, D.J. Energy band structure of BeS, BeSe, and BeTe. Phys. Rev. 1970, 2, 1852. [Google Scholar]
- Al-Douri, Y.; Baaziz, H.; Charifi, Z.; Reshak, A.H. Density functional study of optical properties of beryllium chalcogenides compounds in nickel arsenide B8 structure. Phys. B Condens. Matter 2012, 407, 286–296. [Google Scholar] [CrossRef]
- Guo, L.; Hu, G.; Zhang, S.; Feng, W.; Zhang, Z. Structural, elastic, electronic and optical properties of beryllium chalcogenides BeX (X = S, Se, Te) with zinc-blende structure. J. Alloys Compd. 2013, 561, 16–22. [Google Scholar] [CrossRef]
- Heciri, D.; Beldi, L.; Drablia, S.; Meradji, H.; Derradji, N.E.; Belkhir, H.; Bouhafs, B. First-principles elastic constants and electronic structure of beryllium chalcogenides BeS, BeSe and BeTe. Comput. Mater. Sci. 2007, 38, 609–617. [Google Scholar] [CrossRef]
- Srivastava, G.P.; Tütüncü, H.M.; Günhan, N. First-principles studies of structural, electronic, and dynamical properties of Be chalcogenides. Phys. Rev. B Condens. Matter Mater. Phys. 2004, 70, 1–6. [Google Scholar] [CrossRef]
- Faraji, S.; Mokhtari, A. Ab initio study of the stability and electronic properties of wurtzite and zinc-blende BeS nanowires. Phys. Lett. Sect. A Gen. At. Solid State Phys. 2010, 374, 3348–3353. [Google Scholar] [CrossRef] [Green Version]
- Yadav, P.S.; Yadav, R.K.; Agrawal, S.; Agrawal, B.K. Ab initio study of electronic and optical properties of Be-chalcogenides in GW approximation. Phys. E Low-Dimens. Syst. Nanostructures 2007, 36, 79–85. [Google Scholar] [CrossRef]
- Okoye, C.M.I. Structural, electronic, and optical properties of beryllium monochalcogenides. Eur. Phys. J. B 2004, 39, 5–17. [Google Scholar] [CrossRef]
- Wang, K.L.; Gao, S.P. Phonon dispersions, band structures, and dielectric functions of BeO and BeS polymorphs. J. Phys. Chem. Solids 2018, 118, 242–247. [Google Scholar] [CrossRef]
- Sarkar, R.L.; Chatterjee, S. Electronic energy bands of BeS, BeSe and BeTe. J. Phys. C Solid State Phys. 1977, 10, 57–62. [Google Scholar] [CrossRef]
- Alder, B.J.; Ceperley, D. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45, 566–569. [Google Scholar]
- Vosko, S.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Bagayoko, D.; Zhao, G.L.; Fan, J.D.; Wang, J.T. Ab initio calculations of the electronic structure and optical properties of ferroelectric tetragonal BaTiO3. J. Phys. Condens. Matter 1998, 10, 5645–5655. [Google Scholar] [CrossRef]
- Franklin, L.; Ekuma, C.; Zhao, G.; Bagayoko, D. Bagayoko Density functional theory description of electronic properties of wurtzite zinc oxide. J. Phys. Chem. Solids 2013, 74, 729–736. [Google Scholar] [CrossRef]
- Bagayoko, D.; Franklin, L.; Zhao, G.L. Predictions of electronic, structural, and elastic properties of cubic InN. J. Appl. Phys. 2004, 96, 4297–4301. [Google Scholar] [CrossRef]
- Bagayoko, D. Understanding density functional theory (DFT) and completing it in practice. AIP Adv. 2014, 4, 127104. [Google Scholar] [CrossRef] [Green Version]
- Ayirizia, B.A.; Malozovsky, Y.; Franklin, L.; Bhandari, U.; Bagayoko, D. Ab-Initio Self-Consistent Density Functional Theory Description of Rock-Salt Magnesium Selenide (MgSe). Mater. Sci. Appl. 2020, 11, 401–414. [Google Scholar]
- Bagayoko, D. Understanding the Relativistic Generalization of Density Functional Theory (DFT) and Completing It in Practice. J. Mod. Phys. 2016, 7, 911–919. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, U.; Ayirizia, B.A.; Malozovsky, Y.; Franklin, L.; Bagayoko, D. First principle investigation of electronic, transport, and bulk properties of zinc-blende magnesium sulfide. Electronics 2020, 9, 1791. [Google Scholar] [CrossRef]
- Feibelman, P.J.; Appelbaum, J.A.; Hamann, D.R. Electronic structure of a Ti(0001) film. Phys. Rev. B 1979, 20, 1433–1443. [Google Scholar] [CrossRef]
- Harmon, B.N.; Weber, W.; Hamann, D.R. Total-energy calculations for Si with a first-principles linear-combination-of-atomic-orbitals method. Phys. Rev. B 1982, 25, 1109–1115. [Google Scholar] [CrossRef]
- Ekuma, C.E.; Bagayoko, D. Ab-initio electronic and structural properties of rutile titanium dioxide. Jpn. J. Appl. Phys. 2011, 50, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Narayana, C.; Nesamony, V.; Ruoff, A. Phase transformation of BeS and equation-of-state studies to 96 GPa. Phys. Rev. B Condens. Matter Mater. Phys. 1997, 56, 14338–14343. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Computational Technique | Potential | Band Gap, Eg (eV) |
|---|---|---|
| Pseudopotential Plane Wave (PP-PW) | LDA | 2.911 [5] |
| PP-PW | LDA | 2.75 [6] |
| PP-PW | LDA | 2.816 [8] |
| Empirical Pseudopotential Method | LDA | 2.38 [9] |
| Orthogonalized Plane Wave | LDA | 4.17 [10] |
| Full Potential Linearized Augmented Plane Wave (FP-LAPW) | LDA | 2.828 [11] |
| PP-PW | LDA | 2.912 [12] |
| Augmented plane Waves plus Local orbitals (APW + lo) | LDA | 2.78 [13] |
| PP-PW | LDA | 2.83 [14] |
| FP-LAPW | LSDA | 2.847 [7] |
| PP-PW | GGA | 3.041 [5] |
| PP-PW | GGA | 3.7 [15] |
| PP-PW | GGA | 3.192 [12] |
| PP-PW | GGA | 3.11 [16] |
| FP-LAPW | GGA | 3.141 [11] |
| FP-LAPW | GGA | 3.13 [17] |
| Plane Wave pseudopotential (PW-PP) | GGA | 3.15 [18] |
| APW + lo | Perdew-Burke-Erzerhof (PBE-GGA) | 3.10 [13] |
| PP-PW | Sx-LDA | 4.071 [12] |
| FP-LAPW | EV-GGA | 4.241 [11] |
| Augmented Plane Wave | Hartee-Fock(HF) | 6.10 [19] |
| PW-PP | Quansiparticle Self-Consistent Green’s Function (QPscGW) | 5.27 [18] |
| PW-PP | Green’s Function (GoWo) | 4.62 [18] |
| PP-PW | GoWo | 4.45 [16] |
| Experiment using optical absorption measurement on the BeS platelets | >5.5 [4] |
| Calculation Number | Beryllium (Be2+) | Sulfur (S2−) (1s22s22p6 in Core) | No of Valence Functions | Energy Gap (eV) |
|---|---|---|---|---|
| I | 1s22s02p0 | 3s23p6 | 18 | 7.486 |
| II | 1s22s02p03p0 | 3s23p6 | 24 | 6.345 |
| III | 1s22s02p03p0 | 3s23p64p0 | 30 | 5.438 |
| IV | 1s22s02p03p03s0 | 3s23p64p0 | 32 | 5.406 |
| V | 1s22s02p03p03s0 | 3s23p64p04s0 | 34 | 5.361 |
| L-Point | Γ-Point | X-Point (1–0.35) | X-Point | K-Point |
|---|---|---|---|---|
| 25.451 | 29.334 | 25.824 | 24.420 | 22.099 |
| 17.165 | 29.334 | 15.998 | 16.067 | 14.157 |
| 13.482 | 9.794 | 11.256 | 13.179 | 12.276 |
| 8.463 | 6.784 | 11.256 | 13.179 | 11.816 |
| 8.463 | 6.784 | 10.196 | 7.564 | 10.040 |
| 7.593 | 6.784 | 5.436 | 6.505 | 7.207 |
| −1.121 | 0.000 | −2.215 | −2.775 | −2.299 |
| −1.121 | 0.000 | −2.215 | −2.775 | −4.162 |
| −5.878 | 0.000 | −3.810 | −5.144 | −4.639 |
| −11.970 | −13.585 | −12.215 | −11.424 | −11.439 |
| (a) Me (X-Γ) Longitudinal | Me (X-U) Transverse | Me (X-W) Transverse | |||
|---|---|---|---|---|---|
| 0.743 | 0.317 | 0.313 | |||
| (b) (Γ-L) in (1,1,1) Direction | (Γ-X) in (1,0,0) Direction | (Γ-K) in (1,1,0) Direction | |||
| Mhh | Mlh | Mhh | Mlh | Mhh | Mlh |
| 1.295 | 0.216 | 0.585 | 0.381 | 0.803 | 0.285 |
| Potential | ao (Å) | B | References |
|---|---|---|---|
| LDA | 4.814 | 107.7 | Present |
| LDA | 4.773 | 101.9 | Theory [8] |
| LDA | 4.81 | 93 | Theory [14] |
| LDA | 4.745 | 116 | Theory [6] |
| LDA | 4.800 | 102 | Theory [13] |
| GGA | 4.887 | 92 | Theory [17] |
| PBE-GGA | 4.878 | 93 | Theory [13] |
| Experiment | 4.870 | 105 | Exp [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayirizia, B.A.; Brumfield, J.S.; Malozovsky, Y.; Bagayoko, D. Ground State Properties of the Wide Band Gap Semiconductor Beryllium Sulfide (BeS). Materials 2021, 14, 6128. https://doi.org/10.3390/ma14206128
Ayirizia BA, Brumfield JS, Malozovsky Y, Bagayoko D. Ground State Properties of the Wide Band Gap Semiconductor Beryllium Sulfide (BeS). Materials. 2021; 14(20):6128. https://doi.org/10.3390/ma14206128
Chicago/Turabian StyleAyirizia, Blaise A., Janee’ S. Brumfield, Yuriy Malozovsky, and Diola Bagayoko. 2021. "Ground State Properties of the Wide Band Gap Semiconductor Beryllium Sulfide (BeS)" Materials 14, no. 20: 6128. https://doi.org/10.3390/ma14206128