Melt-Spinning of an Intrinsically Flame-Retardant Polyacrylonitrile Copolymer



Abstract
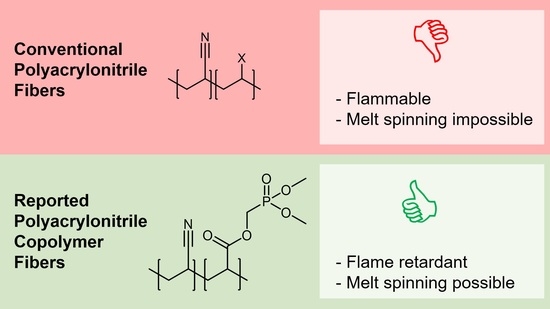
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Synthesis of Dimethyl (Hydroxymethl)phosphonate
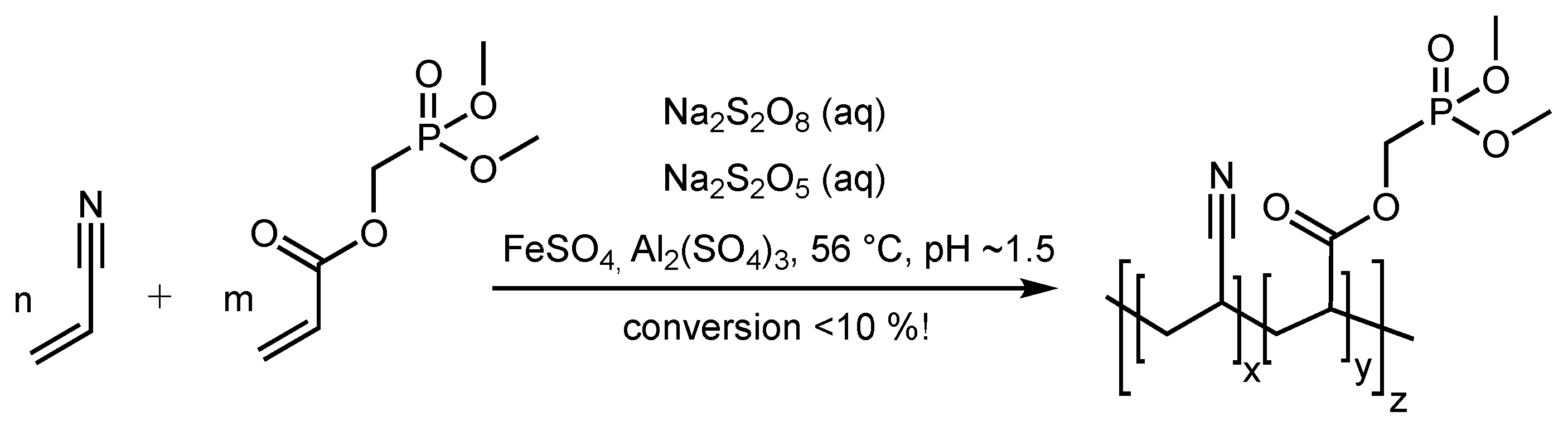
2.3. Synthesis of Dimethyl Phosphonomethylacrylate (DPA)
2.4. Polymerizations
2.5. Calculation of the Copolymerization Parameters
2.6. Drying
2.7. Film Preparation
2.8. Melt Spinning
2.9. Plasticizer Removal
2.10. Characterization
3. Results and Discussion
3.1. Copolymerization Parameters of the Aqueous Precipitation Copolymerization of AN and DPA
3.2. Synthesis and Properties of PAN-copolymers
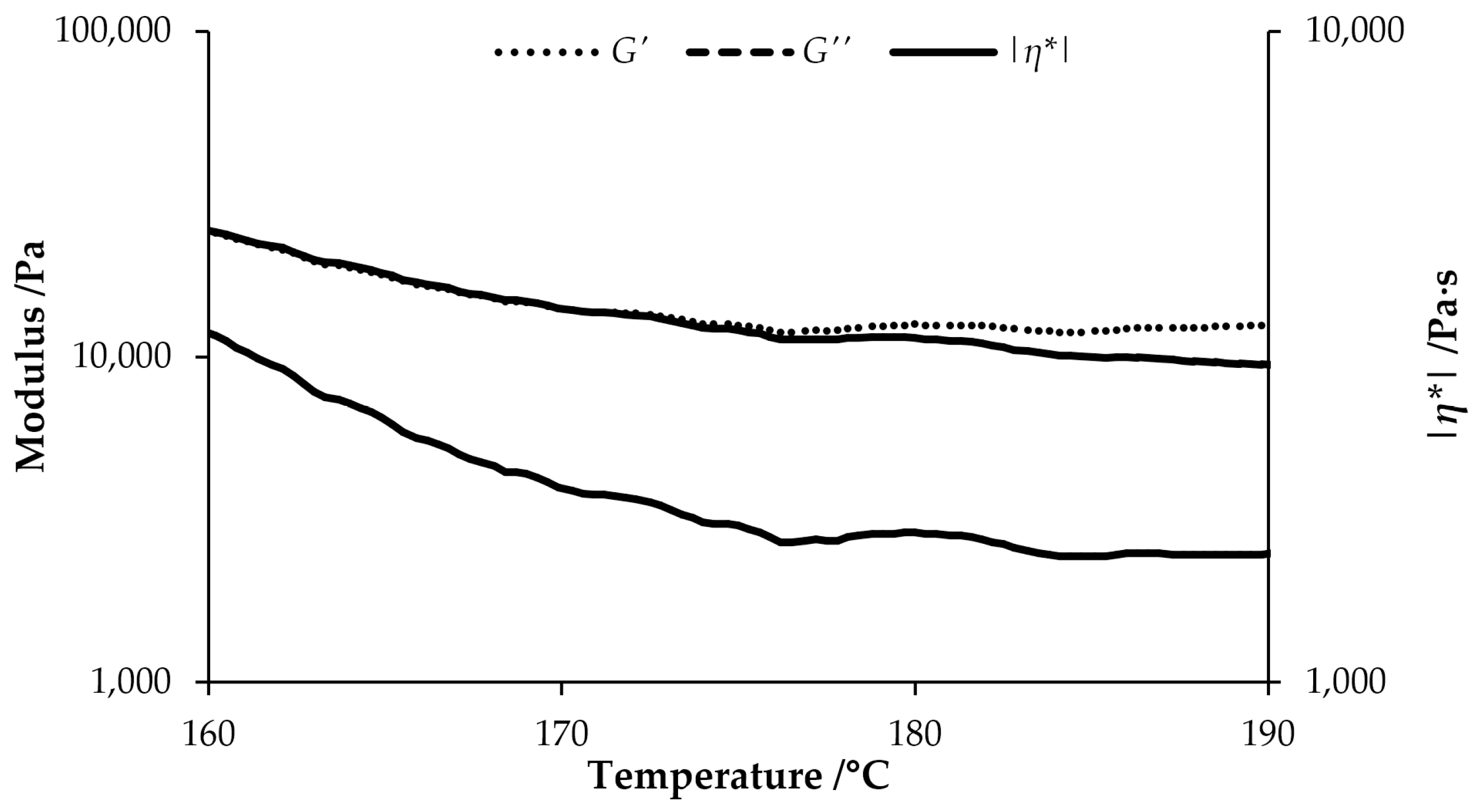
3.3. Rheology and Melt-spinning of a PAN-co-DPA Plasticized with PC
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rein, H. Polyacrylnitril-Fasern Eine neue Gruppe von synthethischen Fasern. Angew. Chem. 1948, 60, 159–161. [Google Scholar] [CrossRef]
- Gupta, B.S.; Afshari, M. 15—Polyacrylonitrile fibers. In Handbook of Properties of Textile and Technical Fibres, 2nd ed.; Bunsell, A.R., Ed.; Woodhead Publishing: Cambridge, UK, 2018; pp. 545–593. [Google Scholar] [CrossRef]
- Spörl, J.M.; Ota, A.; Beyer, R.; Lehr, T.; Müller, A.; Hermanutz, F.; Buchmeiser, M.R. Carbon fibers prepared from tailored reversible-addition-fragmentation transfer copolymerization-derived poly (acrylonitrile)-co-poly (methylmethacrylate). J. Polym. Sci. A1 2014, 52, 1322–1333. [Google Scholar] [CrossRef]
- Frank, E.; Buchmeiser, M.R. (Eds.) Fiber, Films, Resins and Plastics: Carbon Fibers; Springer: Berlin/Heidelberg, Germany, 2015; Volume 1, pp. 306–310. [Google Scholar]
- Frank, E.; Hermanutz, F.; Buchmeiser, M.R. Carbon Fibers: Precursors, Manufacturing and Properties. Macromol. Mater. Eng. 2012, 297, 493–501. [Google Scholar] [CrossRef]
- Frank, E.; Steudle, L.M.; Ingildeev, D.; Sporl, J.M.; Buchmeiser, M.R. Carbon fibers: Precursor systems, processing, structure, and properties. Angew. Chem. Int. Ed. Engl. 2014, 53, 5262–5298. [Google Scholar] [CrossRef]
- Frank, E.; Ingildeev, D.; Buchmeiser, M.R. (Eds.) High-Performance Poly (Acrylonitrile)(PAN)-Based Carbon Fibers and Their Performance Requirements, 1st ed.; Woodhead Publishing Ltd.: Cambridge, UK, 2016; Volume 87, pp. 7–30. [Google Scholar]
- Levchik, S.V.; Weil, E.D. A review of recent progress in phosphorus-based flame retardants. J. Fire Sci. 2006, 24, 345–364. [Google Scholar] [CrossRef]
- Hall, M.E.; Zhang, J.; Richard Horrocks, A. The flammability of polyacrylonitrile and its copolymers III. Effect of flame retardants. Fire Mater. 1994, 18, 231–241. [Google Scholar] [CrossRef]
- Kunststoffe, D.-N. Kunststoffe—Bestimmung des Brennverhaltens durch den Sauerstoff-Index—Teil 1: Allgemeine Anforderungen (ISO 4589-1:2017). In DIN EN ISO 4589-1:2017; DIN Deutsches Institut für Normung e. V.: Berlin, Germany, 2017. [Google Scholar]
- Horrocks, A.; Kandola, B.K.; Davies, P.; Zhang, S.; Padbury, S. Developments in flame retardant textiles—A review. Polym. Degrad. Stab. 2005, 88, 3–12. [Google Scholar] [CrossRef]
- Nametz, R. Flame—Retarding Textile Fibers. Ind. Eng. Chem. 1970, 62, 41–53. [Google Scholar] [CrossRef]
- Karaivanova, S.; Badev, A. Modification of polyacrylonitrile fibers with hydrazine and hydroxylamine in aqueous medium. Angew. Makromol. Chem. 1986, 140, 1–32. [Google Scholar] [CrossRef]
- Kang, Y.Q.; Yang, Y.G.; Li, L.J.; Jia, Z.; Ma, A.R. Structure and Properties of Hydrolyzed Cyclization-Crosslinking Flame-Retardant Polyacrylonitrile Fiber; Trans Tech Publications Ltd.: Stafa-Zurich, Switzerland, 2015; Volume 1120–1121, pp. 576–580. [Google Scholar]
- Zhou, W.; Yan, X.; Liu, P.; Jiang, M.; Xu, J. Flame retardant modification of acrylic fiber with hydrazine hydrate and sodium ions. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Salvio, G.; Gonzato, C.; Tedesco, R.; Marti, J.B. Process for the production of fireproof polyacrylate fibre with a low emission of toxic fumes, uniformly dyed, and acrylic fibres thus obtained. European Patent EP,213,734,2A1, 19 April 2007. [Google Scholar]
- Francalanci, F. Fibre Acriliche. Dalla Crisi Di Mercato A Nuovi Sviluppi. In Proceedings of Assofibre Cires Italia Assembly, City, Italy. Available online: http://www.assofibre.it/assofibre/News.nsf/0/69e68a2f23ebc004c125758d0032683a/$FILE/Montefibre_Francalanci.pdf (accessed on 10 September 2020).
- Vernot, E.; MacEwen, J.; Bruner, R.; Haun, C.; Kinkead, E.; Prentice, D.; HALL, A., III; Schmidt, R.; Eason, R.; Hubbard, G. Long-term inhalation toxicity of hydrazine. Toxicol. Sci. 1985, 5, 1050–1064. [Google Scholar] [CrossRef]
- Yan, X.; Zhou, W.; Zhao, X.; Xu, J.; Liu, P. Preparation, flame retardancy and thermal degradation behaviors of polyacrylonitrile fibers modified with diethylenetriamine and zinc ions. J. Therm. Anal. Calorim. 2016, 124, 719–728. [Google Scholar] [CrossRef]
- Tsai, J.-S. The effect of flame-retardants on the properties of acrylic and modacrylic fibres. J. Mater. Sci. 1993, 28, 1161–1167. [Google Scholar] [CrossRef]
- Johnston, P.; Doyle, E.; Orzel, R. Acrylics: A literature review of thermal decomposition products and toxicity. J. Am. Coll. Toxicol. 1988, 7, 139–200. [Google Scholar] [CrossRef]
- Shaw, S.D.; Blum, A.; Weber, R.; Kannan, K.; Rich, D.; Lucas, D.; Koshland, C.P.; Dobraca, D.; Hanson, S.; Birnbaum, L.S. Halogenated flame retardants: Do the fire safety benefits justify the risks? Rev. Environ. Health 2010, 25, 261. [Google Scholar] [CrossRef] [PubMed]
- Herlinger, H.; Hardtmann, G.; Hermanutz, F.; Schneider, R.; Einsele, U. Herstellung schwer entflammbarer Polyacrylnitrilfasern durch Einspinnen polymerer Phosphorverbindungen. Melliand Textilber. 1991, 72, 353–359. [Google Scholar]
- Hermanutz, F. Herstellung schwer entflammbarer Polyacrylnitrilfasern durch Einspinnen polymerer Phosphorverbindungen. Diploma Thesis, Stuttgart, Germany, 1989. [Google Scholar]
- Liepins, R.; Surles, J.R.; Morosoff, N.; Stannett, V.; Duffy, J.J.; Day, F.H. Localized radiation grafting of flame retardants to polyethylene terephthalate. II. Vinyl phosphonates. J. Appl. Polym. Sci. 1978, 22, 2403–2414. [Google Scholar] [CrossRef]
- Nair, C.P.R.; Clouet, G.; Brossas, J. Copolymerization of diethyl 2-(methacryloyloxy) ethyl phosphate with alkyl acrylates: Reactivity ratios and glass transition temperatures. J. Polym. Sci. A 1988, 26, 1791–1807. [Google Scholar] [CrossRef]
- Tsafack, M.J.; Levalois-Grützmacher, J. Plasma-induced graft-polymerization of flame retardant monomers onto PAN fabrics. Surf. Coat. Technol. 2006, 200, 3503–3510. [Google Scholar] [CrossRef]
- Banks, M.; Ebdon, J.R.; Johnson, M. The flame-retardant effect of diethyl vinyl phosphonate in copolymers with styrene, methyl methacrylate, acrylonitrile and acrylamide. Polymer 1994, 35, 3470–3473. [Google Scholar] [CrossRef]
- Ebdon, J.R.; Hunt, B.J.; Joseph, P.; Wilkie, T.K. Chapter 21 Flame Retardance of Polyacrylonitriles Covalently Modified with Phosphorus- and Nitrogen-Containing Groups. Fire Retard. Polym. 2009, 331–340. [Google Scholar] [CrossRef]
- Herlinger, H.; Veeser, K.; Schaut, G.; Einsele, U. Herstellung schwer entflammbarer Polyacrylnitrilfasern durch Copolymerisation. Textil-Praxis Int. 1979, 9, 1100–1113. [Google Scholar]
- Laoutid, F.; Bonnaud, L.; Alexandre, M.; Lopez-Cuesta, J.-M.; Dubois, P. New prospects in flame retardant polymer materials: From fundamentals to nanocomposites. Mater. Sci. Eng. R 2009, 63, 100–125. [Google Scholar] [CrossRef]
- Babushok, V.; Tsang, W. Inhibitor rankings for alkane combustion. Combust. Flame 2000, 123, 488–506. [Google Scholar] [CrossRef]
- Liu, W.; Chen, D.-Q.; Wang, Y.-Z.; Wang, D.-Y.; Qu, M.-H. Char-forming mechanism of a novel polymeric flame retardant with char agent. Polym. Degrad. Stabil. 2007, 92, 1046–1052. [Google Scholar] [CrossRef]
- Tian, Y.C.; Han, K.Q.; Qin, H.L.; Rong, H.P.; Yan, B.; Wang, D.; Liu, S.P.; Yu, M.H. Rheological Behaviors of Polyacrylonitrile Melt Using Ionic Liquids as a Plasticizer. Adv. Mat. Res. 2012, 476–478, 2151–2157. [Google Scholar] [CrossRef]
- Daumit, G.P.; Ko, Y.S.; Slater, C.R.; Venner, J.G.; Young, C.C. Improvements in the Formation of Melt-Spun Acrylic Fibers. European Patent EP,040,670,9A3, 6 July 1989. [Google Scholar]
- Daumit, G.P.; Ko, Y.S.S.; Christopher, R.; Venner, J.G.; Young, C.C. Melt-Spun Acrylic Fibers Possessing a Highly Uniform Internal Structure which are Particularly Suited for Thermal Conversion to Quality Carbon Fibers. U.S. Patent U,S51,680,04A, 25 August 1988. [Google Scholar]
- Daumit, G.P.; Ko, Y.S.; Slater, C.R.; Venner, J.G.; Young, C.C.; Zwick, M.M. Formation of melt-spun acrylic fibers which are well suited for thermal conversion to high strength carbon fibers. Japan Patent JP,H03,457,08A, 6 July 1989. [Google Scholar]
- König, S.; Kreis, P.; Reinders, L.; Beyer, R.; Wego, A.; Herbert, C.; Steinmann, M.; Frank, E.; Buchmeiser, M.R. Melt spinning of propylene carbonate-plasticized poly(acrylonitrile)-co-poly(methyl acrylate). Polym. Adv. Technol. 2020, 31, 1827–1831. [Google Scholar] [CrossRef] [Green Version]
- Batchelor, B.L.; Mahmood, S.F.; Jung, M.; Shin, H.; Kulikov, O.V.; Voit, W.; Novak, B.M.; Yang, D.J. Plasticization for melt viscosity reduction of melt processable carbon fiber precursor. Carbon 2016, 98, 681–688. [Google Scholar] [CrossRef] [Green Version]
- König, S.; Clauss, M.M.; Giebel, E.; Buchmeiser, M.R. N,N′-Substituted acryloamidines—Novel comonomers for melt-processible poly(acrylonitrile)-based carbon fiber precursors. Polym. Chem. 2019, 10, 4469–4476. [Google Scholar] [CrossRef]
- Porosoff, H. Melt-spinning acrylonitrile polymer fibers. U.S. Patent U,S41,637,70A, 5 February 1973. [Google Scholar]
- Udakhe, J. Melt Processing of Polyacrylonitrile (PAN) Polymers. J. Text. Assoc. 2011, 71, 233–241. [Google Scholar]
- Rangarajan, P.; Yang, J.; Bhanu, V.; Godshall, D.; McGrath, J.; Wilkes, G.; Baird, D. Effect of comonomers on melt processability of polyacrylonitrile. J. Appl. Polym. Sci. 2002, 85, 69–83. [Google Scholar] [CrossRef]
- El Asri, Z.; Chougrani, K.; Negrell-Guirao, C.; David, G.; Boutevin, B.; Loubat, C. An efficient process for synthesizing and hydrolyzing a phosphonated methacrylate: Investigation of the adhesive and anticorrosive properties. J. Polym. Sci. A 2008, 46, 4794–4803. [Google Scholar] [CrossRef]
- Fineman, M.; Ross, S.D. Linear method for determining monomer reactivity ratios in copolymerization. J. Polym. Sci. 1950, 5, 259–262. [Google Scholar] [CrossRef]
- Kelen, T.; Tüdős, F. A new improved linear graphical method for determing copolymerization reactivity ratios. React. Kinet. Catal. Lett. 1974, 1, 487–492. [Google Scholar] [CrossRef]
- Kelen, T.; Tüdõs, F. Analysis of the linear methods for determining copolymerization reactivity ratios. I. A new improved linear graphic method. J. Macromol. Sci. Chem. 1975, 9, 1–27. [Google Scholar] [CrossRef]
- Han, N.; Zhang, X.-X.; Yu, W.-Y.; Gao, X.-Y. Effects of copolymerization temperatures on structure and properties of melt-spinnable acrylonitrile-methyl acrylate copolymers and fibers. Macromol. Res. 2010, 18, 1060–1069. [Google Scholar] [CrossRef]
- Izumi, Z.; Kitagawa, H. Effect of reaction medium on copolymerization of acrylonitrile and methyl acrylate. J. Polym. Sci. A1 1967, 5, 1967–1975. [Google Scholar] [CrossRef]
- Brandrup, J.; Immergut, E.H.; Grulke, E.A.; Abe, A.; Bloch, D.R. Polymer Handbook; Wiley: New York, NY, USA, 1999; Volume 89. [Google Scholar]
- Weil, E.D.; Patel, N.G.; Said, M.; Hirschler, M.M.; Shakir, S. Oxygen index: Correlations to other fire tests. Fire Mater. 1992, 16, 159–167. [Google Scholar] [CrossRef]
- Horrocks, A.R.; Zhang, J.; Hall, M.E. Flammability of polyacrylonitrile and its copolymers II. Thermal behaviour and mechanism of degradation. Polym. Int. 1994, 33, 303–314. [Google Scholar] [CrossRef]
- Sedghi, A.; Farsani, R.E.; Shokuhfar, A. The effect of commercial polyacrylonitrile fibers characterizations on the produced carbon fibers properties. J. Mater. Process. Technol. 2008, 198, 60–67. [Google Scholar] [CrossRef]
- Tsai, J.-S. Modes of stress-strain curve distribution for modacrylic fibres. J. Mater. Sci. 1993, 28, 4841–4845. [Google Scholar] [CrossRef]





{kind=link}
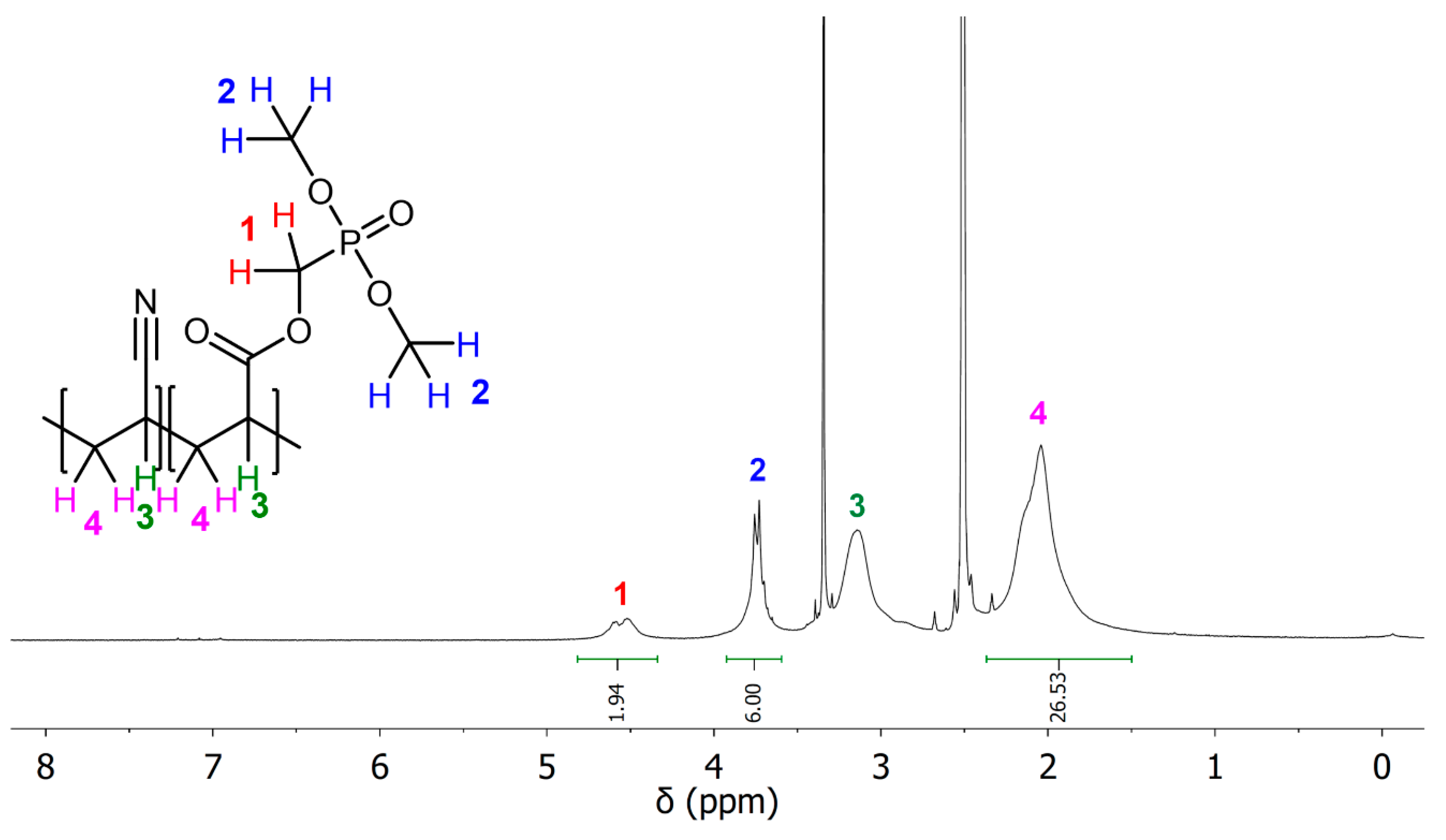{kind=link}
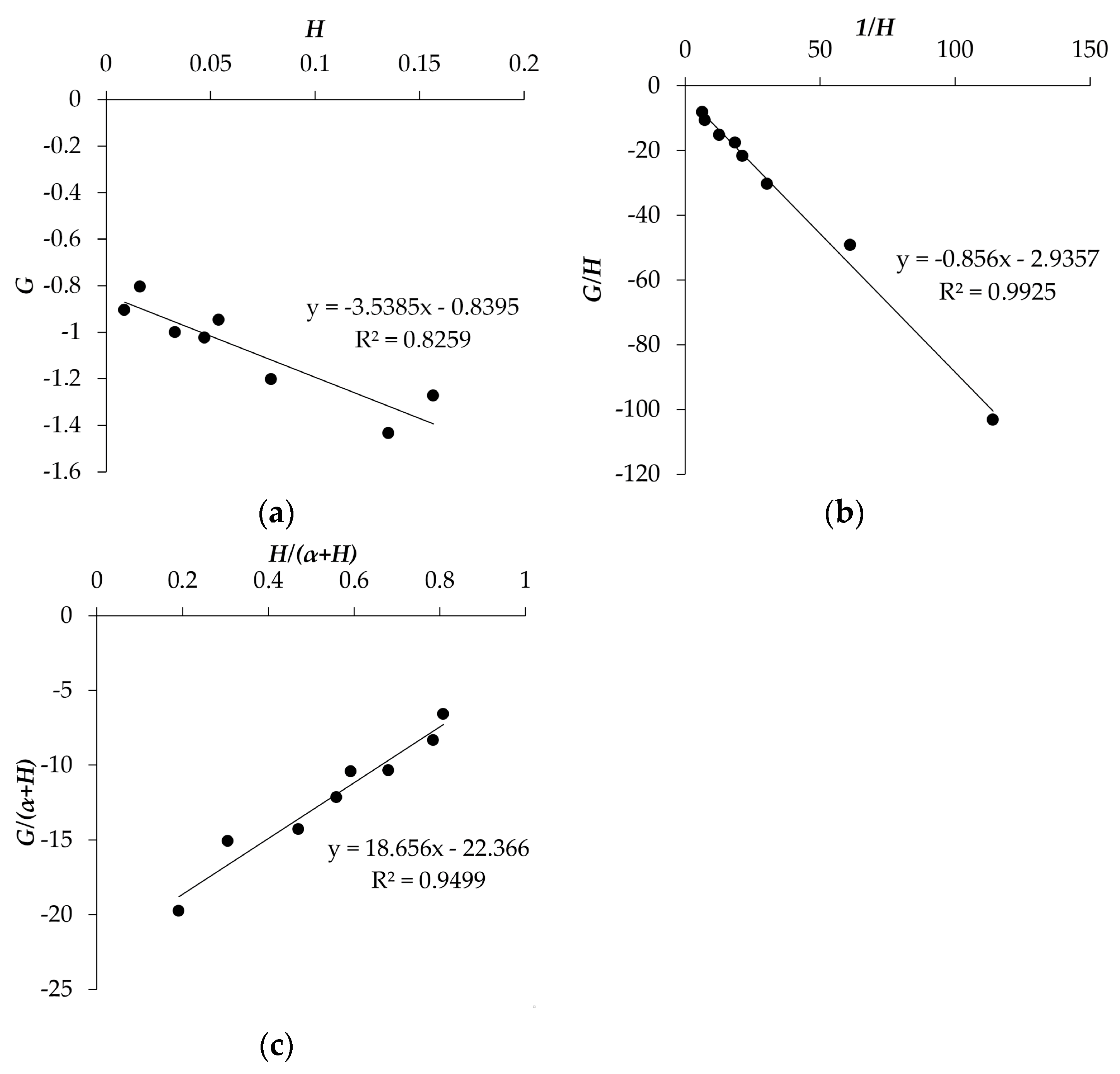{kind=link}
{kind=link}
{kind=link}
| PAN | MAN * /mol% | MDPA * /mol% | MMA * /mol% | P-content Calc. /wt% | P-content Found ** /wt% | mDPA *** /mol% | Đ | Yield /g | X **** /% | |
|---|---|---|---|---|---|---|---|---|---|---|
| PAN F1 | 91.9 | 8.1 | 0 | 3.89 | 3.59 | 7.5 | 43 000 | 5.5 | 34 | 61 |
| PAN F2 | 91.9 | 8.1 | 0 | 3.89 | 3.60 | 7.5 | 35 000 | 5.3 | 30 | 55 |
| PAN F3 | 91.9 | 6.0 | 2.1 | 3 | 2.63 | 5.3 | 51 000 | 4.7 | 28 | 50 |
| PAN F4 | 90.2 | 6.1 | 3.7 | 3 | 2.31 | 4.7 | 57 000 | 4.4 | 7.5 | 67 |
| PAN F5 | 88 | 8.2 | 3.8 | 3.89 | 3.08 | 6.5 | 43 000 | 5.1 | 7.0 | 62 |
| MAN */mol% | MDPA */mol% | mAN **/mol% | mDPA **/mol% | X ***/% |
|---|---|---|---|---|
| 99 | 1 | 99 | 1.0 | 6.4 |
| 98 | 2 | 97.7 | 2.3 | 5.5 |
| 97 | 3 | 97.0 | 3.0 | 7.1 |
| 96 | 4 | 96.0 | 4.0 | 5.1 |
| 95 | 5 | 94.9 | 5.1 | 8.1 |
| 94 | 6 | 95.3 | 4.7 | 6.9 |
| 92 | 8 | 94.5 | 6.6 | 6.6 |
| 90 | 10 | 92.5 | 7.5 | 6.3 |
| Method | rDPA | rAN |
|---|---|---|
| Fineman and Ross | 0.84 ± 0.06 | 3.5 ± 0.7 |
| Inverted Fineman and Ross | 0.86 ± 0.03 | 2.9 ± 1.5 |
| Kelen and Tüdõs | 0.83 ± 0.06 | 3.7 ± 1.4 |
| Average | 0.84 ± 0.05 | 3.4 ± 1.2 |
| PAN | P-content Found/wt% | LOI (Film) | Tg/°C | Tdeg/°C |
|---|---|---|---|---|
| PAN F1 | 3.59 | 26.4 | 99 | 240 |
| PAN F2 | 3.60 | 26.5 | 98 | 248 |
| PAN F3 | 2.63 | 23.2 | 99 | 250 |
| PAN F4 | 2.31 | 22.2 | 99 | 255 |
| PAN F5 | 3.08 | 25.1 | 95 | 239 |
| Sample | Possible Spinning Temperature/°C | tan δ at 170 °C | tan δ at 175 °C | |η*| at 170 °C/Pa⋅s | |η*| at 175 °C/Pa⋅s |
|---|---|---|---|---|---|
| PAN F1/PC | - | 0.38 | 0.38 | 7600 | 7600 |
| PAN F2/PC | - | 0.34 | 0.32 | 7100 | 7800 |
| PAN F3/PC | - | n.d.* | n.d.* | n.d.* | n.d.* |
| PAN F4/PC | - | 0.47 | 0.42 | 11,600 | 12,000 |
| PAN F5/PC | 170(-175?) | 1.0 | 0.97 | 1970 | 1740 |
| Plasticizer Removed | Diameter /μm | Elongation /% | Tenacity /cN/tex | Tenacity /MPa | Young’s Modulus /cN/tex | Young’s Modulus /GPa |
|---|---|---|---|---|---|---|
| no | 49 ± 9 | 15.5 ± 2.5 | 9 ± 3 | 100 ± 40 | 80 ± 30 | 1.0 ± 0.4 |
| yes | 44 ± 13 | 19 ± 5 | 17 ± 4 | 195 ± 40 | 440 ± 60 | 5.2 ± 0.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
König, S.; Kreis, P.; Herbert, C.; Wego, A.; Steinmann, M.; Wang, D.; Frank, E.; Buchmeiser, M.R. Melt-Spinning of an Intrinsically Flame-Retardant Polyacrylonitrile Copolymer. Materials 2020, 13, 4826. https://doi.org/10.3390/ma13214826
König S, Kreis P, Herbert C, Wego A, Steinmann M, Wang D, Frank E, Buchmeiser MR. Melt-Spinning of an Intrinsically Flame-Retardant Polyacrylonitrile Copolymer. Materials. 2020; 13(21):4826. https://doi.org/10.3390/ma13214826
Chicago/Turabian StyleKönig, Simon, Philipp Kreis, Christian Herbert, Andreas Wego, Mark Steinmann, Dongren Wang, Erik Frank, and Michael R. Buchmeiser. 2020. "Melt-Spinning of an Intrinsically Flame-Retardant Polyacrylonitrile Copolymer" Materials 13, no. 21: 4826. https://doi.org/10.3390/ma13214826