Design of Zeolite-Covalent Organic Frameworks for Methane Storage



Abstract
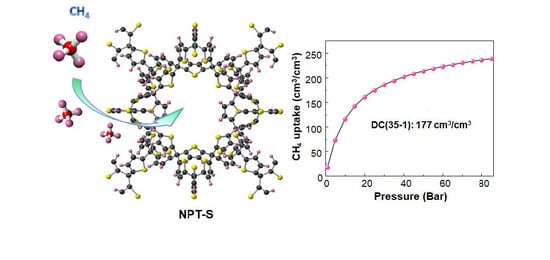
:
1. Introduction
2. Design Strategy and Methodologies
3. Results and Discussion
3.1. Screening ZCOFs for Methane Storage
3.2. Adsorption of CH4
3.3. Methane Delivery Capacity
3.4. CH4 Adsorption Sites
3.5. The Formation Energy of JST-S and NPT-S
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cote, A.P.; El-Kaderi, H.M.; Furukawa, H.; Hunt, J.R.; Yaghi, O.M. Reticular synthesis of microporous and mesoporous 2D covalent organic frameworks. J. Am. Chem. Soc. 2007, 129, 12914–12915. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Eddaoudi, M.; O’Keeffe, M.; Yaghi, O.M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 1999, 402, 276–279. [Google Scholar] [CrossRef] [Green Version]
- Eddaoudi, M.; Kim, J.; Rosi, N.; Vodak, D.; Wachter, J.; O’Keeffe, M.; Yaghi, O.M. Systematic design of pore size and functionality in isoreticular MOFs and their application in methane storage. Science 2002, 295, 469–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Düren, T.; Sarkisov, L.; Yaghi, O.M.; Snurr, R.Q. Design of new materials for methane storage. Langmuir 2004, 20, 2683–2689. [Google Scholar] [CrossRef]
- Mendoza-Cortés, J.L.; Han, S.S.; Furukawa, H.; Yaghi, O.M.; Goddard III, W.A. Adsorption mechanism and uptake of methane in covalent organic frameworks: Theory and experiment. J. Phys. Chem. A 2010, 114, 10824–10833. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.-B.; He, Y.; Li, P.; Wang, H.; Zhou, W.; Chen, B. Multifunctional porous hydrogen-bonded organic framework materials. Chem. Soc. Rev. 2019, 48, 1362–1389. [Google Scholar] [CrossRef]
- Park, K.S.; Ni, Z.; Côté, A.P.; Choi, J.Y.; Huang, R.; Uribe-Romo, F.J.; Chae, H.K.; O’Keeffe, M.; Yaghi, O.M. Exceptional chemical and thermal stability of zeolitic imidazolate frameworks. Proc. Natl. Acad. Sci. USA 2006, 103, 10186–10191. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Kravtsov, V.C.; Larsen, R.; Eddaoudi, M. Molecular building blocks approach to the assembly of zeolite-like metal–organic frameworks (ZMOFs) with extra-large cavities. Chem. Commun. 2006, 1488–1490. [Google Scholar] [CrossRef]
- Eddaoudi, M.; Sava, D.F.; Eubank, J.F.; Adil, K.; Guillerm, V. Zeolite-like metal–organic frameworks (ZMOFs): Design, synthesis, and properties. Chem. Soc. Rev. 2015, 44, 228–249. [Google Scholar] [CrossRef] [Green Version]
- Cote, A.P.; Benin, A.I.; Ockwig, N.W.; O’Keeffe, M.; Matzger, A.J.; Yaghi, O.M. Porous, crystalline, covalent organic frameworks. Science 2005, 310, 1166–1170. [Google Scholar] [CrossRef] [Green Version]
- Dogru, M.; Sonnauer, A.; Gavryushin, A.; Knochel, P.; Bein, T. A covalent organic framework with 4 nm open pores. Chem. Commun. 2011, 47, 1707–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Zhou, J.; Wang, W.; Wang, H. Progress in Synthesis and Application of Covalent Organic Frameworks. Ekoloji 2019, 28, 4369–4378. [Google Scholar]
- Guan, X.; Chen, F.; Fang, Q.; Qiu, S. Design and applications of three dimensional covalent organic frameworks. Chem. Soc. Rev. 2020, 49, 1357–1384. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Li, B.; Zhu, R.; Pang, H. Design and synthesis of covalent organic frameworks towards energy and environment fields. Chem. Eng. J. 2019, 355, 602–623. [Google Scholar] [CrossRef]
- Song, Y.; Sun, Q.; Aguila, B.; Ma, S. Opportunities of covalent organic frameworks for advanced applications. Adv. Sci. 2019, 6, 1801410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, M.; Lan, Y.; Qin, Z.; Zhong, C. Computation-ready, experimental covalent organic framework for methane delivery: Screening and material design. J. Phys. Chem. C 2018, 122, 13009–13016. [Google Scholar] [CrossRef]
- Sharma, R.K.; Yadav, P.; Yadav, M.; Gupta, R.; Rana, P.; Srivastava, A.; Zbořil, R.; Varma, R.S.; Antonietti, M.; Gawande, M.B. Recent development of covalent organic frameworks (COFs): Synthesis and catalytic (organic-electro-photo) applications. Mater. Horiz. 2020, 7, 411–454. [Google Scholar] [CrossRef]
- Liu, J.; Wang, N.; Ma, L. Recent Advances in Covalent Organic Frameworks for Catalysis. Chem. Asian J. 2019, 15, 338–351. [Google Scholar] [CrossRef]
- Ascherl, L.; Evans, E.W.; Gorman, J.; Orsborne, S.; Bessinger, D.; Bein, T.; Friend, R.H.; Auras, F. Perylene-Based Covalent Organic Frameworks for Acid Vapor Sensing. J. Am. Chem. Soc. 2019, 141, 15693–15699. [Google Scholar] [CrossRef]
- Gao, Q.; Li, X.; Ning, G.-H.; Leng, K.; Tian, B.; Liu, C.; Tang, W.; Xu, H.-S.; Loh, K.P. Highly photoluminescent two-dimensional imine-based covalent organic frameworks for chemical sensing. Chem. Commun. 2018, 54, 2349–2352. [Google Scholar] [CrossRef]
- El-Kaderi, H.M.; Hunt, J.R.; Mendoza-Cortés, J.L.; Côté, A.P.; Taylor, R.E.; O’Keeffe, M.; Yaghi, O.M. Designed synthesis of 3D covalent organic frameworks. Science 2007, 316, 268–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, J.R.; Doonan, C.J.; LeVangie, J.D.; Côté, A.P.; Yaghi, O.M. Reticular synthesis of covalent organic borosilicate frameworks. J. Am. Chem. Soc. 2008, 130, 11872–11873. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Ko, N.; Go, Y.B.; Aratani, N.; Choi, S.B.; Choi, E.; Yazaydin, A.Ö.; Snurr, R.Q.; O’Keeffe, M.; Kim, J. Ultrahigh porosity in metal-organic frameworks. Science 2010, 329, 424–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burchell, T.; Rogers, M. Low pressure storage of natural gas for vehicular applications. SAE Trans. 2000, 109, 2242–2246. [Google Scholar]
- Mendoza-Cortes, J.L.; Pascal, T.A.; Goddard III, W.A. Design of covalent organic frameworks for methane storage. J. Phys. Chem. A 2011, 115, 13852–13857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Zhao, J.; Yan, T. Methane uptakes in covalent organic frameworks with double halogen substitution. J. Phys. Chem. C 2015, 119, 2010–2014. [Google Scholar] [CrossRef]
- Wu, H.; Zhou, W.; Yildirim, T. High-capacity methane storage in metal−organic frameworks M2 (dhtp): The important role of open metal sites. J. Am. Chem. Soc. 2009, 131, 4995–5000. [Google Scholar] [CrossRef]
- Ma, S.; Sun, D.; Simmons, J.M.; Collier, C.D.; Yuan, D.; Zhou, H.-C. Metal-organic framework from an anthracene derivative containing nanoscopic cages exhibiting high methane uptake. J. Am. Chem. Soc. 2008, 130, 1012–1016. [Google Scholar] [CrossRef]
- Konstas, K.; Osl, T.; Yang, Y.; Batten, M.; Burke, N.; Hill, A.J.; Hill, M.R. Methane storage in metal organic frameworks. J. Mater. Chem 2012, 22, 16698–16708. [Google Scholar] [CrossRef]
- Schoedel, A.; Ji, Z.; Yaghi, O.M. The role of metal–organic frameworks in a carbon-neutral energy cycle. Nat. Energy 2016, 1, 1–13. [Google Scholar] [CrossRef]
- Zhao, J.; Yan, T. Effects of substituent groups on methane adsorption in covalent organic frameworks. RSC Adv. 2014, 4, 15542–15551. [Google Scholar] [CrossRef]
- Martin, R.L.; Simon, C.M.; Smit, B.; Haranczyk, M. In silico design of porous polymer networks: High-throughput screening for methane storage materials. J. Am. Chem. Soc. 2014, 136, 5006–5022. [Google Scholar] [CrossRef]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.; Goddard III, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Willems, T.F.; Rycroft, C.H.; Kazi, M.; Meza, J.C.; Haranczyk, M. Algorithms and tools for high-throughput geometry-based analysis of crystalline porous materials. Microporous Mesoporous Mater. 2012, 149, 134–141. [Google Scholar] [CrossRef]
- Düren, T.; Millange, F.; Férey, G.; Walton, K.S.; Snurr, R.Q. Calculating geometric surface areas as a characterization tool for metal− organic frameworks. J. Phys. Chem. C 2007, 111, 15350–15356. [Google Scholar] [CrossRef]
- Gupta, A.; Chempath, S.; Sanborn, M.J.; Clark, L.A.; Snurr, R.Q. Object-oriented programming paradigms for molecular modeling. Mol. Simul 2003, 29, 29–46. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Bo, J.; Wu, L. Molecular simulation of CH 4/CO 2/H 2 O competitive adsorption on low rank coal vitrinite. Phys. Chem. Chem. Phys. 2017, 19, 17773–17788. [Google Scholar] [CrossRef]
- Martin, M.G.; Siepmann, J.I. Transferable potentials for phase equilibria. 1. United-atom description of n-alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. [Google Scholar]
- Zarkova, L.; Hohm, U.; Damyanova, M. Comparison of Lorentz–Berthelot and Tang–Toennies Mixing Rules Using an Isotropic Temperature-Dependent Potential Applied to the Thermophysical Properties of Binary Gas Mixtures of CH4, CF4, SF6, and C(CH3)4 with Ar, Kr, and Xe. Int. J. Thermophys. 2004, 25, 1775–1798. [Google Scholar] [CrossRef]
- Düren, T.; Bae, Y.-S.; Snurr, R.Q. Using molecular simulation to characterise metal–organic frameworks for adsorption applications. Chem. Soc. Rev. 2009, 38, 1237–1247. [Google Scholar] [CrossRef]
- Pascale, F.; Zicovich-Wilson, C.M.; Orlando, R.; Roetti, C.; Ugliengo, P.; Dovesi, R. Vibration frequencies of Mg3Al2Si3O12 pyrope. An ab initio study with the CRYSTAL code. J. Phys. Chem. B 2005, 109, 6146–6152. [Google Scholar] [PubMed]
- Pham, H.Q.; Mai, T.; Pham-Tran, N.-N.; Kawazoe, Y.; Mizuseki, H.; Nguyen-Manh, D. Engineering of band gap in metal–organic frameworks by functionalizing organic linker: A systematic density functional theory investigation. J. Phys. Chem. C 2014, 118, 4567–4577. [Google Scholar] [CrossRef]
- Li, B.; Wen, H.-M.; Zhou, W.; Xu, J.Q.; Chen, B. Porous metal-organic frameworks: Promising materials for methane storage. Chem 2016, 1, 557–580. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, H.; Yaghi, O.M. Storage of hydrogen, methane, and carbon dioxide in highly porous covalent organic frameworks for clean energy applications. J. Am. Chem. Soc. 2009, 131, 8875–8883. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Atom | ε/kb (K) | σ (Å) | ref |
|---|---|---|---|---|
| CH4 | - | 148.0 | 3.73 | [38] |
| ZCOFs | C | 52.8 | 3.43 | [33] |
| H | 22.1 | 2.57 | - | |
| S | 137.9 | 3.59 | - | |
| O | 30.2 | 3.12 | - | |
| N | 34.7 | 3.26 | - |
| ZCOFs | Sacc (m2/g) | Dpore (Å) | Vpore (cm3/g) | Total Uptake at 35 bar (cm3/cm3) | Delivery Capacity 35–1 bar (cm3/cm3) | Total Uptake at 65 bar (cm3/cm3) | Delivery Capacity 65–5.8 bar |
|---|---|---|---|---|---|---|---|
| BOZ-S | 2579 | 16.1 | 0.516 | 191 | 174 | 227 | 152 |
| JSR-N | 2865 | 12.8 | 0.543 | 197 | 174 | 231 | 150 |
| JSR-O | 2702 | 12.4 | 0.489 | 187 | 170 | 221 | 151 |
| JSR-S | 2480 | 13.6 | 0.612 | 179 | 168 | 220 | 164 |
| JST-S | 2615 | 10.7 | 0.433 | 179 | 169 | 230 | 183 |
| NPT-S | 2548 | 19.5 | 0.502 | 194 | 177 | 227 | 145 |
| OBW-N | 2920 | 13.8 | 0.451 | 191 | 166 | 221 | 134 |
| OBW-S | 2734 | 15.7 | 0.557 | 191 | 174 | 226 | 148 |
| RWY-N | 3437 | 24.2 | 1.212 | 156 | 147 | 206 | 161 |
| RWY-O | 3273 | 22.8 | 1.055 | 158 | 149 | 208 | 163 |
| RWY-S | 3209 | 26.5 | 1.419 | 135 | 129 | 189 | 156 |
| Bulk CH4 | - | - | - | 34 | 33 | 66 | 61 |
| ZCOFs | Sacc (m2/g) | Dpore (Å) | Vpore (cm3/g) | CH4 Uptake (cm3/cm3) | CH4 Delivery (cm3/cm3) | Ref |
|---|---|---|---|---|---|---|
| COF-102-Ant | 2720 | - | 0.75 | 215 | 180 | [25] |
| COF-103-Eth-trans | 4920 | - | 1.36 | 206 | 192 | [25] |
| COF-102-1,4-2I | - | - | - | - | 181 | [26] |
| COF-102-I | - | - | - | 176 | 169 | [31] |
| COF-102-Cl | - | - | - | 169 | 165 | [31] |
| COF-1 | 750 | 9 | 0.30 | 55 | - | [44] |
| COF-5 | 1670 | 27 | 1.07 | 73 | - | [44] |
| COF-6 | 750 | 9 | 0.32 | 101 | - | [44] |
| COF-8 | 1350 | 16 | 0.69 | 85 | - | [44] |
| COF-10 | 1760 | 32 | 1.44 | 53 | - | [44] |
| COF-102 | 3620 | 12 | 1.55 | 113 | - | [44] |
| COF-103 | 3530 | 12 | 1.54 | 105 | - | [44] |
| NPT-S | 2548 | 19.5 | 0.502 | 194 | 177 | This work |
| ZCOF | Symmetry | Atom/Cell | Lattice Parameter (Å) | ∆H (kJ/mol) |
|---|---|---|---|---|
| JST-S | Pa-3 | 720 | 27.3204 | −34,823 × 103 |
| NPT-S | Pm-3 | 540 | 25.0125 | −8705 × 103 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Do, H.H.; Kim, S.Y.; Le, Q.V.; Pham-Tran, N.-N. Design of Zeolite-Covalent Organic Frameworks for Methane Storage. Materials 2020, 13, 3322. https://doi.org/10.3390/ma13153322
Do HH, Kim SY, Le QV, Pham-Tran N-N. Design of Zeolite-Covalent Organic Frameworks for Methane Storage. Materials. 2020; 13(15):3322. https://doi.org/10.3390/ma13153322
Chicago/Turabian StyleDo, Ha Huu, Soo Young Kim, Quyet Van Le, and Nguyen-Nguyen Pham-Tran. 2020. "Design of Zeolite-Covalent Organic Frameworks for Methane Storage" Materials 13, no. 15: 3322. https://doi.org/10.3390/ma13153322