Hydrogen Trapping in bcc Iron


 , and
, and
Abstract
:1. Introduction
2. Computational Details
2.1. Electronic Structure and Total Energy Calculations
2.2. Structure Models
2.2.1. Bulk
2.2.2. Interfaces
2.2.3. Dislocations
3. Methodology
3.1. Solution Energies
3.2. Hydrogen Trapping at Defects
3.3. Effect of H on the Bulk Cohesive Strength
3.4. Effect of Trapping on GB Cohesive Strength
3.5. Determination of H Concentration from Segregation Energies
4. Results
4.1. Pure Fe
4.1.1. Bulk
Ground State Properties of the Bulk Fe
Vacancy
Dislocation
4.1.2. Interface
Grain Boundary and Free Surface
4.2. Iron + Hydrogen
4.2.1. Hydrogen Trapping in the Bulk
Hydrogen Solubility in Fe Lattice
Hydrogen Trapping at A Vacancy
Hydrogen Trapping at a Dislocation
4.2.2. Interfaces
Hydrogen Trapping at GB
Effect of H on the Bulk and GB Cohesion
5. Discussion
5.1. Trap Hierarchy at 0 K
5.2. Traps at Finite Temperatures
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nagumo, M. Fundamentals of Hydrogen Embrittlement; Springer: Singapore, 2016; ISBN 9789811001611. [Google Scholar]
- Bhadeshia, H.K.D.H. Prevention of hydrogen embrittlement in steels. ISIJ Int. 2016, 56, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Robertson, I.M.; Sofronis, P.; Nagao, A.; Martin, M.L.; Wang, S.; Gross, D.W.; Nygren, K.E. Hydrogen embrittlement understood. Metall. Mater. Trans. A Phys. Metall. Mater. Sci. 2015, 46, 2323–2341. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, M.; Kameda, J.; Ebihara, K.-I.; Itakura, M.; Kaburaki, H. Mobile effect of hydrogen on intergranular decohesion of iron: First-principles calculations. Philos. Mag. 2012, 92, 1349–1368. [Google Scholar] [CrossRef]
- Hickel, T.; Nazarov, R.; McEniry, E.J.; Leyson, G.; Grabowski, B.; Neugebauer, J. Ab initio based understanding of the segregation and diffusion mechanisms of hydrogen in steels. Jom 2014, 66, 1399–1405. [Google Scholar] [CrossRef]
- Geng, W.-T.; Freeman, A.J.; Olson, G.B.; Tateyama, Y.; Ohno, T. Hydrogen-promoted grain boundary embrittlement and vacancy activity in metals: Insights from ab initio total energy calculatons. Mater. Trans. 2005, 46, 756–760. [Google Scholar] [CrossRef] [Green Version]
- Pérez Escobar, D.; Depover, T.; Duprez, L.; Verbeken, K.; Verhaege, M. Combined thermal desorption spectroscopy, differential scanning calorimetry, scanning electron microscopy and X-ray diffraction study of hydrogen trapping in cold deformed TRIP steel. Acta Mater. 2012, 60, 2593–2605. [Google Scholar] [CrossRef]
- Pérez Escobar, D.; Duprez, L.; Atrens, A.; Verbeken, K. Influence of experimental parameters on thermal desorption spectroscopy measurements during evaluation of hydrogen trapping. J. Nucl. Mater. 2014, 450, 32–41. [Google Scholar] [CrossRef]
- Geng, W.T.; Freeman, A.J. Embrittling and strengthening effects of hydrogen, boron, and phosphorus on a Σ 5 nickel grain boundary. Phys. Rev. B 1999, 60, 7149–7155. [Google Scholar] [CrossRef]
- Zhong, L.; Wu, R.; Freeman, A.J.; Olson, G.B. Charge transfer mechanism of hydrogen-induced intergranular embrittlement of iron. Phys. Rev. B Condens. Matter Mater. Phys. 2000, 62, 13938–13941. [Google Scholar] [CrossRef]
- Mishin, Y.; Asta, M.; Li, J. Atomistic modeling of interfaces and their impact on microstructure and properties. Acta Mater. 2010, 58, 1117–1151. [Google Scholar] [CrossRef] [Green Version]
- Mirzaev, D.A.; Mirzoev, A.A.; Okishev, K.Y.; Verkhovykh, A.V. Hydrogen–vacancy interaction in bcc iron: Ab initio calculations and thermodynamics. Mol. Phys. 2014, 112, 1745–1754. [Google Scholar] [CrossRef]
- Mirzaev, D.A.; Mirzoev, A.A.; Okishev, K.Y.; Verkhovykh, A.V. Ab initio modelling of the interaction of H interstitials with grain boundaries in bcc Fe. Mol. Phys. 2016, 114, 1502–1512. [Google Scholar] [CrossRef]
- McEniry, E.J.; Hickel, T.; Neugebauer, J.J. Hydrogen behaviour at twist {110} grain boundaries in α-Fe. Philos. Trans. A Math. Phys. Eng. Sci. 2017, 375, 20160402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEniry, E.J.; Dey, P.; Hickel, T.; Neugebauer, J. Atomistic Modelling of Cosegregation at Structural Defects in Steels. SteelyHydrogen. Available online: http://steelyhydrogen2018proc.be/articles/atomistic-modelling-of-cosegregation-at-structural-defects-in-steels/25 (accessed on 15 May 2020).
- Zhao, Y.; Lu, G. QM/MM study of dislocation—Hydrogen/helium interactions in α-Fe. Model. Simul. Mater. Sci. Eng. 2011, 19, 065004. [Google Scholar] [CrossRef]
- Itakura, M.; Kaburaki, H.; Yamaguchi, M.; Okita, T. The effect of hydrogen atoms on the screw dislocation mobility in bcc iron: A first-principles study. Acta Mater. 2013, 61, 6857–6867. [Google Scholar] [CrossRef]
- Tateyama, Y.; Ohno, T. Stability and clusterization of hydrogen-vacancy complexes in α-Fe: An ab initio study. Phys. Rev. B 2003, 67, 1–2. [Google Scholar] [CrossRef]
- Wang, F.; Shang, J.; Li, J.; Wang, C. The effects of boron and hydrogen on the embrittlement of polycrystalline Ni3Al. Intermetallics 2000, 8, 589–593. [Google Scholar] [CrossRef]
- Qi, Y.; Hector, L.G. Hydrogen effect on adhesion and adhesive transfer at aluminum/diamond interfaces. Phys. Rev. B 2003, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Siegl, W.; Ecker, J.; Klarner, G.; Kloesch, G.; Mori, A.; Drexler, G.; Winter, H. Hydrogen Trapping in Heat Treated and Deformed Armco Iron; NACE International: Houston, TX, USA, 2019. [Google Scholar]
- Mceniry, E.J.; Hickel, T.; Neugebauer, J. Atomistic modelling of light-element co-segregation at structural defects in iron. Procedia Struct. Integr. 2018, 13, 1099–1104. [Google Scholar] [CrossRef]
- Lu, T.; Xu, Y.P.; Pan, X.D.; Zhou, H.S.; Ding, F.; Yang, Z.; Niu, G.J.; Luo, G.N.; Li, X.C.; Gao, F. Atomistic study of hydrogen behavior around dislocations in α iron. J. Nucl. Mater. 2018, 510, 219–228. [Google Scholar] [CrossRef]
- Tateyama, Y.; Ohno, T. Atomic-scale effects of hydrogen in iron toward hydrogen embrittlement: Ab-initio study. ISIJ Int. 2003, 43, 573–578. [Google Scholar] [CrossRef]
- Razumovskiy, V.I.; Divinski, S.V.; Romaner, L. Acta Materialia Solute segregation in Cu: DFT vs. Experiment. Acta Mater. 2018, 147, 122–132. [Google Scholar] [CrossRef]
- Scheiber, D.; Razumovskiy, V.I.; Puschnig, P.; Pippan, R.; Romaner, L. Ab initio description of segregation and cohesion of grain boundaries in W-25 at.% Re alloys. Acta Mater. 2015, 88, 180–189. [Google Scholar] [CrossRef]
- He, B.; Xiao, W.; Hao, W.; Tian, Z. First-principles investigation into the effect of Cr on the segregation of multi-H at the Fe Σ3 (1 1 1) grain boundary. J. Nucl. Mater. 2013, 441, 301–305. [Google Scholar] [CrossRef]
- Lu, T.; Niu, G.; Xu, Y.; Wang, J.; An, Z.; Liu, H.; Zhou, H.; Ding, F.; Luo, G.; Li, X. Molecular dynamics study of the diffusion properties of H in Fe with point defects. Fusion Eng. Des. 2016, 113, 340–345. [Google Scholar] [CrossRef]
- Lv, G.; Zhang, M.; Zhang, H.; Su, Y. Hydrogen diffusion and vacancy clusterization in iron. Int. J. Hydrogen Energy 2018, 43, 15378–15385. [Google Scholar] [CrossRef]
- Kimizuka, H.; Ogata, S. Slow diffusion of hydrogen at a screw dislocation core in α-iron. Phys. Rev. B Condens. Matter Mater. Phys. 2011, 84, 1–6. [Google Scholar] [CrossRef]
- Teus, S.M.; Mazanko, V.F.; Olive, J.M.; Gavriljuk, V.G. Grain boundary migration of substitutional and interstitial atoms in α-iron. Acta Mater. 2014, 69, 105–113. [Google Scholar] [CrossRef]
- Jiang, D.E.; Carter, E.A. Diffusion of interstitial hydrogen into and through bcc Fe from first principles. Phys. Rev. B Condens. Matter Mater. Phys. 2004, 70, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, J.; Fullea, J.; Andrade, C.; De Andres, P.L. Hydrogen in α -iron: Stress and diffusion. Phys. Rev. B Condens. Matter Mater. Phys. 2008, 78, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Svoboda, J.; Fischer, F.D. Modelling for hydrogen diffusion in metals with traps revisited. Acta Mater. 2012, 60, 1211–1220. [Google Scholar] [CrossRef]
- Fischer, F.D.; Mori, G.; Svoboda, J. Modelling the influence of trapping on hydrogen permeation in metals. Corros. Sci. 2013, 76, 382–389. [Google Scholar] [CrossRef]
- Drexler, A.; Depover, T.; Verbeken, K.; Ecker, W. Model-based interpretation of thermal desorption spectra of Fe-C-Ti alloys. J. Alloys Compd. 2019, 789, 647–657. [Google Scholar] [CrossRef]
- Drexler, A.; Depover, T.; Leitner, S.; Verbeken, K.; Ecker, W. Microstructural based hydrogen diffusion and trapping models applied to Fe–C-X alloys. J. Alloys Compd. 2020, 154057. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar]
- Kresse, G.; Hafner, J. Ab. initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal—Amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillonin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallograph. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Birch, F. Finite elastic strain of cubic crystals. Phys. Rev. B 1947, 71, 809–824. [Google Scholar] [CrossRef]
- Murnaghan, F.D. The compressibility of media under extreme pressures. Proc. Natl. Acad. Sci. USA 1944, 30, 244–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razumovskiy, V.I.; Scheiber, D.; Razumovskii, I.M.; Butrim, V.N.; Trushnikova, A.S.; Varlamova, S.B.; Beresnev, A.G. New Cr-Ni-base alloy for high-temperature applications designed on the basis of first principles calculations. Adv. Condens. Matter Phys. 2018, 9383981. [Google Scholar] [CrossRef] [Green Version]
- Ventelon, L.; Willaime, F. Core structure and Peierls potential of screw dislocations in α-Fe from first principles: Cluster versus dipole approaches. J. Comput. Mater. Des. 2007, 14, 85. [Google Scholar] [CrossRef]
- Segall, E.; Strachan, A.; Goddard, W.; Ismail-Beigi, S.; Arias, A. Ab initio and finite-temperature molecular dynamics studies of lattice resistance in tantalum. Phys. Rev. B Condens. Matter Mater. Phys. 2003, 68, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Bulatov, V.V.; Chang, J.; Li, J.; Yip, S. Anisotropic elastic interactions of a periodic dislocation array. Phys. Rev. Lett. 2001, 86, 5727–5730. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, C.Z.; Chang, J.P.; Cai, W.; Bulatov, V.V.; Ho, K.M.; Yip, S. Core energy and Peierls stress of a screw dislocation in bcc molybdenum: A periodic-cell tight-binding study. Phys. Rev. B Condens. Matter Mater. Phys. 2004, 70, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wurster, S.; Motz, C.; Romaner, L.; Ambrosch-Draxl, C.; Pippan, R. Dislocation-core symmetry and slip planes in tungsten alloys: Ab initio calculations and microcantilever bending experiments. Acta Mater. 2012, 60, 748–758. [Google Scholar] [CrossRef]
- Subramaniam, D.; Libisch, F.; Li, Y.; Pauly, C.; Geringer, V.; Reiter, R.; Mashoff, T.; Liebmann, M.; Burgdörfer, J.; Busse, C.; et al. Wave-function mapping of graphene quantum dots with soft confinement. Phys. Rev. Lett. 2012, 108, 46801. [Google Scholar] [CrossRef] [PubMed]
- Romaner, L.; Razumovskiy, V.I.; Pippan, R. Core polarity of screw dislocations in Fe-Co alloys. Philos. Mag. Lett. 2014, 94, 334–341. [Google Scholar] [CrossRef]
- Razumovskii, I.M.; Ruban, A.V.; Razumovskiy, V.I.; Logunov, A.V.; Larionov, V.N.; Ospennikova, O.G.; Poklad, V.A.; Johansson, B. New generation of Ni-based superalloys designed on the basis of first-principles calculations. Mater. Sci. Eng. A 2008, 497, 18–24. [Google Scholar] [CrossRef]
- Finnis, M.W. The theory of metal-ceramic interfaces. J. Phys. Condens. Matter 1996, 8, 5811. [Google Scholar] [CrossRef]
- Razumovskiy, V.I.; Lozovoi, A.Y.; Razumovskii, I.M. First-principles-aided design of a new Ni-base superalloy: Influence of transition metal alloying elements on grain boundary and bulk cohesion. Acta Mater. 2015, 82, 369–377. [Google Scholar] [CrossRef]
- Rice, J.R.; Wang, J.S. Embrittlement of interfaces by solute segregation. Mater. Sci. Eng. A 1989, 107, 23–40. [Google Scholar] [CrossRef]
- Sutton, A.; Balluffi, R. Interfaces in Crystalline Materials; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The materials project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1. [Google Scholar] [CrossRef] [Green Version]
- Hayward, E.; Fu, C.C. Interplay between hydrogen and vacancies in α-Fe. Phys. Rev. B Condens. Matter Mater. Phys. 2013, 87, 1–14. [Google Scholar] [CrossRef]
- Haas, P.; Tran, F.; Blaha, P. Calculation of the lattice constant of solids with semilocal functionals. Phys. Rev. B Condens. Matter Mater. Phys. 2009, 79, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Forderer, K.I.; Doring, K.; Gladisch, M.; Haas, N.; Herlach, D.; Major, J.; Mundinger, H.; Rosenkranz, J.; Schafer, W.; Schimmele, L.; et al. μ+sr study of vacancies in thermal equilibrium in ferromagnets. Hyperfine Interact. 1986, 31, 81–86. [Google Scholar] [CrossRef]
- Razumovskiy, V.I.; Ruban, A.V.; Korzhavyi, P.A. Effect of Temperature on the Elastic Anisotropy of Pure Fe and Fe_{0.9}Cr_{0.1} Random Alloy. Phys. Rev. Lett. 2011, 107, 205504. [Google Scholar] [CrossRef]
- Ruban, A.V.; Razumovskiy, V.I. Spin-wave method for the total energy of paramagnetic state. Phys. Rev. B Condens. Matter Mater. Phys. 2012, 85, 1–10. [Google Scholar] [CrossRef]
- Razumovskiy, V.I.; Ruban, A.V.; Korzhavyi, P.A. First-principles study of elastic properties of Cr- and Fe-rich Fe-Cr alloys. Phys. Rev. B Condens. Matter Mater. Phys. 2011, 84, 1–8. [Google Scholar] [CrossRef]
- Razumovskiy, V.I.; Reyes-Huamantinco, A.; Puschnig, P.; Ruban, A.V. Effect of thermal lattice expansion on the stacking fault energies of fcc Fe and Fe75Mn25 alloy. Phys. Rev. B 2016, 93, 1–8. [Google Scholar] [CrossRef]
- Rayne, J.A.; Chandrasekhar, B.S. Elastic constants of iron from 4.2 to 300 °K. Phys. Rev. 1961, 122, 1714–1716. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics; Wiley: Hoboken, NJ, USA, 1996; ISBN 9780471490210. [Google Scholar]
- Söderlind, P.; Yang, L.; Moriarty, J.; Wills, J. First-principles formation energies of monovacancies in bcc transition metals. Phys. Rev. B 2000, 61, 2579–2586. [Google Scholar] [CrossRef]
- Domain, C.; Becquart, C. Ab initio calculations of defects in Fe and dilute Fe-Cu alloys. Phys. Rev. B 2001, 65, 1–14. [Google Scholar] [CrossRef]
- Olsson, P.; Domain, C.; Wallenius, J. Ab initio study of Cr interactions with point defects in bcc Fe. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 75, 1–12. [Google Scholar] [CrossRef]
- Momida, H.; Asari, Y.; Nakamura, Y.; Tateyama, Y.; Ohno, T. Hydrogen-enhanced vacancy embrittlement of grain boundaries in iron. Phys. Rev. B Condens. Matter Mater. Phys. 2013, 88, 31–33. [Google Scholar] [CrossRef]
- Matsumoto, R.; Sera, M.; Miyazaki, N. Hydrogen concentration estimation in metals at finite temperature using first-principles calculations and vibrational analysis. Comput. Mater. Sci. 2014, 91, 211–222. [Google Scholar] [CrossRef]
- Zu, X.T.; Yang, L.; Gao, F.; Peng, S.M.; Heinisch, H.L.; Long, X.G.; Kurtz, R.J. Properties of helium defects in bcc and fcc metals investigated with density functional theory. Phys. Rev. B Condens. Matter Mater. Phys. 2009, 80, 1–6. [Google Scholar] [CrossRef]
- Ohnuma, T.; Soneda, N.; Iwasawa, M. First-principles calculations of vacancy-solute element interactions in body-centered cubic iron. Acta Mater. 2009, 57, 5947–5955. [Google Scholar] [CrossRef]
- Counts, W.A.; Wolverton, C.; Gibala, R. First-principles energetics of hydrogen traps in α-Fe: Point defects. Acta Mater. 2010, 58, 4730–4741. [Google Scholar] [CrossRef]
- Paxton, A.T.; Elsässer, C. Electronic structure and total energy of interstitial hydrogen in iron: Tight-binding models. Phys. Rev. B Condens. Matter Mater. Phys. 2010, 82, 13–15. [Google Scholar] [CrossRef] [Green Version]
- De Schepper, L.; Segers, D.; Dorikens-Vanpraet, L.; Dorikens, M.; Knuyt, G.; Stals, L.M.; Moser, P. Positron annihilation on pure and carbon-doped -iron in thermal equilibrium. Phys. Rev. B 1983, 27, 5257–5269. [Google Scholar] [CrossRef]
- Robertson, I.M.; Lillig, D.; Ferreira, P.J. Revealing the Fundamental Processes Controlling Hydrogen Embrittlmente. In Effects of Hydrogen on Materials; ASM International: Cleveland, OH, USA, 2009; pp. 22–37. [Google Scholar]
- Liang, Y.; Sofronis, P.; Aravas, N. On the effect of hydrogen on plastic instabilities in metals. Acta Mater. 2003, 51, 2717–2730. [Google Scholar] [CrossRef]
- Leyson, G.P.M.; Grabowski, B.; Neugebauer, J. Multiscale description of dislocation induced nano-hydrides. Acta Mater. 2015, 89, 50–59. [Google Scholar] [CrossRef]
- Lynch, S.P. A fractographic study of hydrogen-assisted cracking and liquid-metal embrittlement in nickel. J. Mater. Sci. 1986, 21, 692. [Google Scholar] [CrossRef]
- Barrera, O.; Bombac, D.; Chen, Y.; Daff, T.D.; Galindo-Nava, E.; Gong, P.; Haley, D.; Horton, R.; Katzarov, I.; Kermode, J.R.; et al. Understanding and mitigating hydrogen embrittlement of steels: A review of experimental, modelling and design progress from atomistic to continuum. J. Mater. Sci. 2018, 53, 6251–6290. [Google Scholar] [CrossRef] [Green Version]
- Woodward, C.; Rao, S.I. Flexible Ab Initio Boundary Conditions: Simulating Isolated Dislocations in bcc Mo and Ta. Phys. Rev. Lett. 2002, 88, 4. [Google Scholar] [CrossRef]
- Romaner, L.; Ambrosch-Draxl, C.; Pippan, R. Effect of rhenium on the dislocation core structure in tungsten. Phys. Rev. Lett. 2010, 104, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Vitek, V. Theory of the core structures of dislocations in body-centred-cubic metals. Cryst. Latt. Defects 1974, 5, 1. [Google Scholar]
- Ventelon, L.; Willaime, F.; Clouet, E.; Rodney, D. Ab initio investigation of the Peierls potential of screw dislocations in bcc Fe and W. Acta Mater. 2013, 61, 3973–3985. [Google Scholar] [CrossRef]
- Frederiksen, S.L.; Jacobsen, K.W. Density functional theory studies of screw dislocation core structures in bcc metals. Phil. Mag. 2003, 83, 365. [Google Scholar] [CrossRef]
- Wan, L.; Geng, W.T.; Ishii, A.; Du, J.P.; Mei, Q.; Ishikawa, N.; Kimizuka, H.; Ogata, S. Hydrogen embrittlement controlled by reaction of dislocation with grain boundary in alpha-iron. Int. J. Plast. 2019, 112, 206–219. [Google Scholar] [CrossRef] [Green Version]
- Iannuzzi, M.; Barnoush, A.; Johnsen, R. Materials and corrosion trends in offshore and subsea oil and gas production. NPJ Mater. Degrad. 2017, 1. [Google Scholar] [CrossRef]
- Wang, S.; Martin, M.L.; Sofronis, P.; Ohnuki, S.; Hashimoto, N.; Robertson, I.M. Hydrogen-induced intergranular failure of iron. Acta Mater. 2014, 69, 275–282. [Google Scholar] [CrossRef]
- Kim, S.M.; Buyers, W.J.L. Vacancy formation energy in iron by positron annihilation. J. Phys. F Met. Phys. 1978, 8, L103–L108. [Google Scholar] [CrossRef]
- Maier, K.; Metz, H.; Herlach, D.; Schaefer, H.E. High temperature positron annihilation experiments in BCC metals. J. Nucl. Mater. 1978, 69, 589–592. [Google Scholar] [CrossRef]
- Schaefer, H.E.; Maier, K.; Weller, M.; Herlach, D.; Seeger, A.; Diehl, J. Vacancy formation in iron investigated by positron annihilation in thermal equilibrium. Scr. Metall. 1977, 11, 803–809. [Google Scholar] [CrossRef]
- Seeger, A. Lattice vacancies in high-purity α-iron. Phys. Status Solidi 1998, 167, 289–311. [Google Scholar] [CrossRef]
- Shin, Y.K.; Kwak, H.; Zou, C.; Vasenkov, A.V.; Van Duin, A.C.T. Development and validation of a ReaxFF reactive force field for Fe/Al/Ni alloys: Molecular dynamics study of elastic constants, diffusion, and segregation. J. Phys. Chem. A 2012, 116, 12163–12174. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.K.; Tanaka, S.; Shiihara, Y.; Kohyama, M. Ab initio study of symmetrical tilt grain boundaries in bcc Fe: structural units, magnetic moments, interfacial bonding, local energy and local stress. J. Phys. Condens. Matter 2013, 25, 135004. [Google Scholar] [CrossRef] [PubMed]
- Mirzoev, A.A.; Mirzaev, D.A.; Verkhovykh, A.V. Hydrogen-vacancy interactions in ferromagnetic and paramagnetic bcc iron: Ab initio calculations. Phys. Status Solidi Basic Res. 2015, 252, 1966–1970. [Google Scholar] [CrossRef]
- Wang, J.; Madsen, G.K.H.; Drautz, R. Grain boundaries in bcc-Fe: A density-functional theory and tight-binding study. Model. Simul. Mater. Sci. Eng. 2018, 26. [Google Scholar] [CrossRef]
- Wachowicz, E.; Ossowski, T.; Kiejna, A. Cohesive and magnetic properties of grain boundaries in bcc Fe with Cr additions. Phys. Rev. B Condens. Matter Mater. Phys. 2010, 81, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Scheiber, D.; Pippan, R.; Puschnig, P.; Romaner, L. Ab initio calculations of grain boundaries in bcc metals. Model. Simul. Mater. Sci. Eng. 2016, 24, 35013. [Google Scholar] [CrossRef]
- Tyson, W.R.; Miller, W.A. Surface free energies of solid metals: Estimation from liquid surface tension measurements. Surf. Sci. 1977, 62, 267–276. [Google Scholar] [CrossRef]
- Boer, F.R.; Boom, R.; Mattens, W.C.M.; Miedema, A.R.; Niessen, A.K. Cohesion in Metals; North Holland: Amsterdam, The Netherlands, 1988. [Google Scholar]
- Spencer, M.J.S.; Hung, A.; Snook, I.K.; Yarovsky, I. Density functional theory study of the relaxation and energy of iron surfaces. Surf. Sci. 2002, 513, 389–398. [Google Scholar] [CrossRef]
- Jiang, D.E.; Carter, E.A. First principles assessment of ideal fracture energies of materials with mobile impurities: Implications for hydrogen embrittlement of metals. Acta Mater. 2004, 52, 4801–4807. [Google Scholar] [CrossRef]
- Kuopanportti, P.; Hayward, E.; Fu, C.C.; Kuronen, A.; Nordlund, K. Interatomic Fe-H potential for irradiation and embrittlement simulations. Comput. Mater. Sci. 2016, 111, 525–531. [Google Scholar] [CrossRef] [Green Version]
- Mishin, Y.; Sorensen, M.R.; Voter, A.F. Calculation of point-defect entropy in metals. Philos. Mag. A 2001, 81, 2591–2612. [Google Scholar] [CrossRef] [Green Version]
- Hirth, J.P. Effects of hydrogen on the properties of iron and steel. Metall. Trans. A 1980, 11, 861–890. [Google Scholar] [CrossRef]
- Christmann, K. Interaction of hydrogen with solid surfaces. Surf. Sci. Rep. 1988, 9, 1–163. [Google Scholar] [CrossRef]
- Wipf, H. Solubility and diffusion of hydrogen in pure metals and alloys. Phys. Scr. 2001, T94, 43–51. [Google Scholar] [CrossRef]
- Du, Y.A.; Ismer, L.; Rogal, J.; Hickel, T.; Neugebauer, J.; Drautz, R. First-principles study on the interaction of H interstitials with grain boundaries in α- and γ-Fe. Phys. Rev. B Condens. Matter Mater. Phys. 2011, 84, 1–13. [Google Scholar] [CrossRef]
- Ohsawa, K.; Eguchi, K.; Watanabe, H.; Yamaguchi, M.; Yagi, M. Configuration and binding energy of multiple hydrogen atoms trapped in monovacancy in bcc transition metals. Phys. Rev. B Condens. Matter Mater. Phys. 2012, 85, 1–8. [Google Scholar] [CrossRef]
- Besenbacher, F.; Myers, S.M.; Nordlander, P.; Nørskov, J.K. Multiple hydrogen occupancy of vacancies in Fe. J. Appl. Phys. 1987, 61, 1788–1794. [Google Scholar] [CrossRef]
- Myers, S.M.; Follstaedt, D.M.; Besenbacher, F.; Bøttiger, J. Trapping and surface permeation of deuterium in He-implanted Fe. J. Appl. Phys. 1982, 53, 8734–8744. [Google Scholar] [CrossRef]
- Ramasubramaniam, A.; Itakura, M.; Carter, E.A. Interatomic potentials for hydrogen in α–iron based on density functional theory. Phys. Rev. B 2009, 79, 174101. [Google Scholar] [CrossRef]
- Choo, W.Y.; Lee, J.Y. Thermal analysis of trapped hydrogen in pure iron. Met. Trans. A. 1982, 13, 135–140. [Google Scholar] [CrossRef]
- Bernstein, I.M. The effect of hydrogen on the deformation of iron. Scr. Metall. 1974, 8, 343–349. [Google Scholar] [CrossRef]
- Pressouyre, G.M. A classification of hydrogen traps in steel. Metall. Trans. A 1979, 10, 1571–1573. [Google Scholar] [CrossRef]
- Hagi, H.; Hayashi, Y. Diffusion coefficients of hydrogen in carbon steels between 278 and 318 K and hydrogen trapping effect of interface between cementite and ferrite. Jpn. Inst. Met. 1993, 57, 864–869. [Google Scholar] [CrossRef] [Green Version]
- Miodownik, G.M.; Achar, B.S. Proceedings of International Conference on “I’Hydrogene dans les Metaux”; Sciences-Industries: Paris, France, 1972. [Google Scholar]
- Gibala, R. Internal friction in hydrogen-charged iron. Trans. Metall. Soc. AIME 1967, 239, 1574–1585. [Google Scholar]
- Gibala, R.; Counts, W.A.; Wolverton, C. The hydrogen cold work peak in BCC iron: Revisited, with first principles calculations and implications for hydrogen embrittlement. Mater. Res. 2018, 21. [Google Scholar] [CrossRef]
- Voronoi, G. Nouvelles applications des paramètres continus à la théorie des formes quadratiques. J. Reine Angew. Math. 1908, 133, 97–178. [Google Scholar] [CrossRef]
- Ventelon, L.; Lüthi, B.; Clouet, E.; Proville, L.; Legrand, B.; Rodney, D.; Willaime, F. Dislocation core reconstruction induced by carbon segregation in bcc iron. Phys. Rev. B Condens. Matter Mater. Phys. 2015, 91, 1–5. [Google Scholar] [CrossRef] [Green Version]
- McLean, D. Grain Boundaries in Metals; Clarendon Press: Oxford, UK, 1957. [Google Scholar]










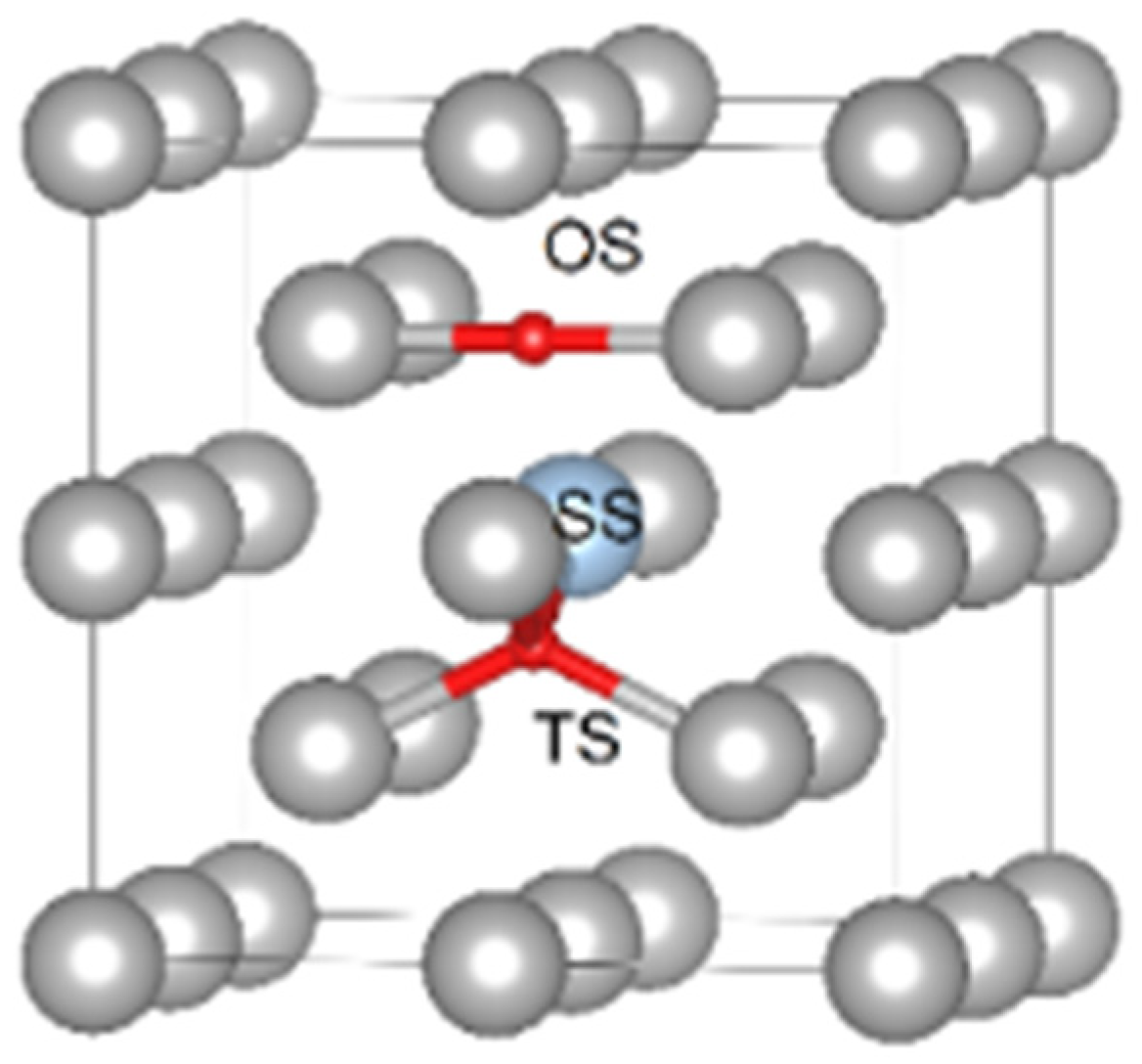{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | a, Å | B, GPa | µ, µB |
|---|---|---|---|
| This work, PBE | 2.831 | 181 | 2.19 |
| Material project DFT [61] | 2.847 | 182 | 2.33 |
| Sanchez08 DFT, PBE [33] | 2.815 | 175 | 2.25 |
| Hayward13 DFT,PBE [62] | 2.834 | 174 | 2.20 |
| Rayne61 Exp. 3.2 K [69] | - | 173 | - |
| Söderlind00 DFT [71] | 2.836 | 195 | - |
| Haas09 DFT [63] | 2.833 | - | - |
| Haas09 Exp. [63] | 2.853 | - | - |
| Characteristic | Defect Type | This Work | DFT Studies | Experimental |
|---|---|---|---|---|
| Formation energy, e | Vacancy | 2.02 | 1.93, 1.95, 2.01 [72], 1.86, 2.06, 2.16 [75], 2.14 [76], 2.15 [73,74,77], 2.17 [77,78], 2.37 [18], 2.39 [79] | 1.4 [94], 1.5 [95], 1.6 [96], 1.61–1.75 [97], 1.7 [64], 2.00 [80] |
| FS energy, J/m2 | (111) FS | 2.67 | 2.52 [106], 2.69 [4], 2.69 [98], 2.65 [74], 2.71, 3.23 [103] | 2.42 [104], 2.48 [105] |
| (012) FS | 2.44 | |||
| (100) FS | 2.94 | 2.55, 3.06 [103], 2.29 [106] | ||
| GB energy, J/m2 | Σ3 (111) [1,2,3,4,5,6,7,8,9,10] GB | 1.60 | 1.57 [102], 1.52 [4], 1.66 [74], 1.46 [100], 1.61 [99], 1.57 [101], 1.79 [103] | - |
| Σ5 (012) [100] | 1.60 | 2.00 [102], 1.64 [101], 1.83 [13] | - | |
| Σ5 (100) [001] | 2.01 | 2.12 [101], 2.20 [103] | - | |
| Work of separation, J/m2 | Σ3 (111) [1,2,3,4,5,6,7,8,9,10] | 3.76 | 3.86 [4], 3.65 [74], 3.78 [102], 4.60 [103] | - |
| Σ5 (012) [100] | 2.88 | 3.19 [102] | - | |
| Σ5 (100) [001] | 3.86 | 3.90 [103] | - |
| Type of H Site | Solution Energy, eV | ||
|---|---|---|---|
| This Work | Theoretical | Experimental | |
| Interstitital tetrahedral | 0.23 (4 × 4 × 4 cell) 0.22 (dilute limit) | 0.19 [107], 0.21 [78], 0.23 [62], 0.27 [79] | 0.30 (0.20) [110,111], 0.28 (0.18) [112] |
| Interstitital octahedral | 0.37 (4 × 4 × 4 cell) | 0.26 [62], 0.32 [107], 0.34 [78], 0.35 [79] | - |
| substitutional | 2.54 (4 × 4 × 4 cell) | 2.53 [108], 2.61 [78] | - |
| Type of Defect | Literature Data at 0 K | Literature Data at 0 K + ZPE (Defect +H) | Method | This Work | This Work + ZPE | Experimental Data |
|---|---|---|---|---|---|---|
| Vacancy | DFT, PBE | |||||
| H1V | –0.69 [117], −0.57 [114], −0.6 [12], −0.5 [62] | −0.56 [24], −0.62 [62] | DFT PW91 [12] DFT PBE [13,14,22,57,104] | −0.58 | −0.70 (−0.12) [62] | −0.63 [115] |
| H2V | −0.61 [12,24,114], -0.54 [62] | −0.65 [62] | −0.63 | −0.74 (−0.11) [62] | ||
| H3V | −0.40 [24,114], −0.39 [12],−0.34 [62] | −0.38 [62] | −0.39 | −0.43 (−0.04) [62] | −0.43 [115] | |
| H4V | −0.27 [24], −0.36 [114], −0.37 [12], −0.30 [62] | −0.35 [62] | ||||
| H5V | −0.33 [24], −0.32 [114], −0.31 [12], −0.27 [62] | −0.27 [62] | ||||
| H6V | 0.02 [24], 0.01 [114], 0.043 [62] | −0.045 [62] | ||||
| GB | ||||||
| Tilt Σ3 (111) | −0.39 [107] | −0.58 [27] | DFT PBE | −0.47 | −0.57 (−0.1) [27] | −0.18 [118] −0.28 [119] −0.61 [120] |
| Tilt Σ5 (012) | −0.81 [107] | −0.42 | ||||
| Tilt Σ5 (013) | −0.43 [107] | |||||
| Tilt Σ9 (1/2 11) | −0.29 [15] | TB | ||||
| Tilt Σ13 (1/3 11) | −0.27 [15] | |||||
| Tilt Σ17 (1/4 11) | −0.32 [15] | |||||
| Twist Σ3(110) | −0.26 [17] | |||||
| Twist Σ5 (100) | - | −0.57 | ||||
| Twist Σ9(110) | −0.68 [17] | |||||
| Twist Σ11(110) | −0.83 [17] | |||||
| Twist Σ17(110) | −0.95 [17] | |||||
| Dislocation | ||||||
| Edge | −0.47[16] | QM/MM | −0.28 [118] −0.20 [121] −0.31 [122] −0.25 [123] | |||
| Screw1/2 <111> | −0.27 [19,20], 0.2 to −0.3 [124], −0.26 [23] | −0.32 [17] | QM/MM [19], DFT, PBE [20,125], MD [23] | −0.21 | −0.26 (−0.05 [17]) | |
| Mixed <111> | ~−0.3 [124] | - | DFT PBE | −0.37 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kholtobina, A.S.; Pippan, R.; Romaner, L.; Scheiber, D.; Ecker, W.; Razumovskiy, V.I. Hydrogen Trapping in bcc Iron. Materials 2020, 13, 2288. https://doi.org/10.3390/ma13102288
Kholtobina AS, Pippan R, Romaner L, Scheiber D, Ecker W, Razumovskiy VI. Hydrogen Trapping in bcc Iron. Materials. 2020; 13(10):2288. https://doi.org/10.3390/ma13102288
Chicago/Turabian StyleKholtobina, Anastasiia S., Reinhard Pippan, Lorenz Romaner, Daniel Scheiber, Werner Ecker, and Vsevolod I. Razumovskiy. 2020. "Hydrogen Trapping in bcc Iron" Materials 13, no. 10: 2288. https://doi.org/10.3390/ma13102288