Anisotropic-Cyclicgraphene: A New Two-Dimensional Semiconducting Carbon Allotrope




Abstract
:1. Introduction
2. Computational Methods
2.1. Prediction of Two-Dimensional Materials
2.2. Ab Initio Computations
2.3. Finite Temperature Stability-Molecular Dynamics Calculations
2.4. Structural Properties
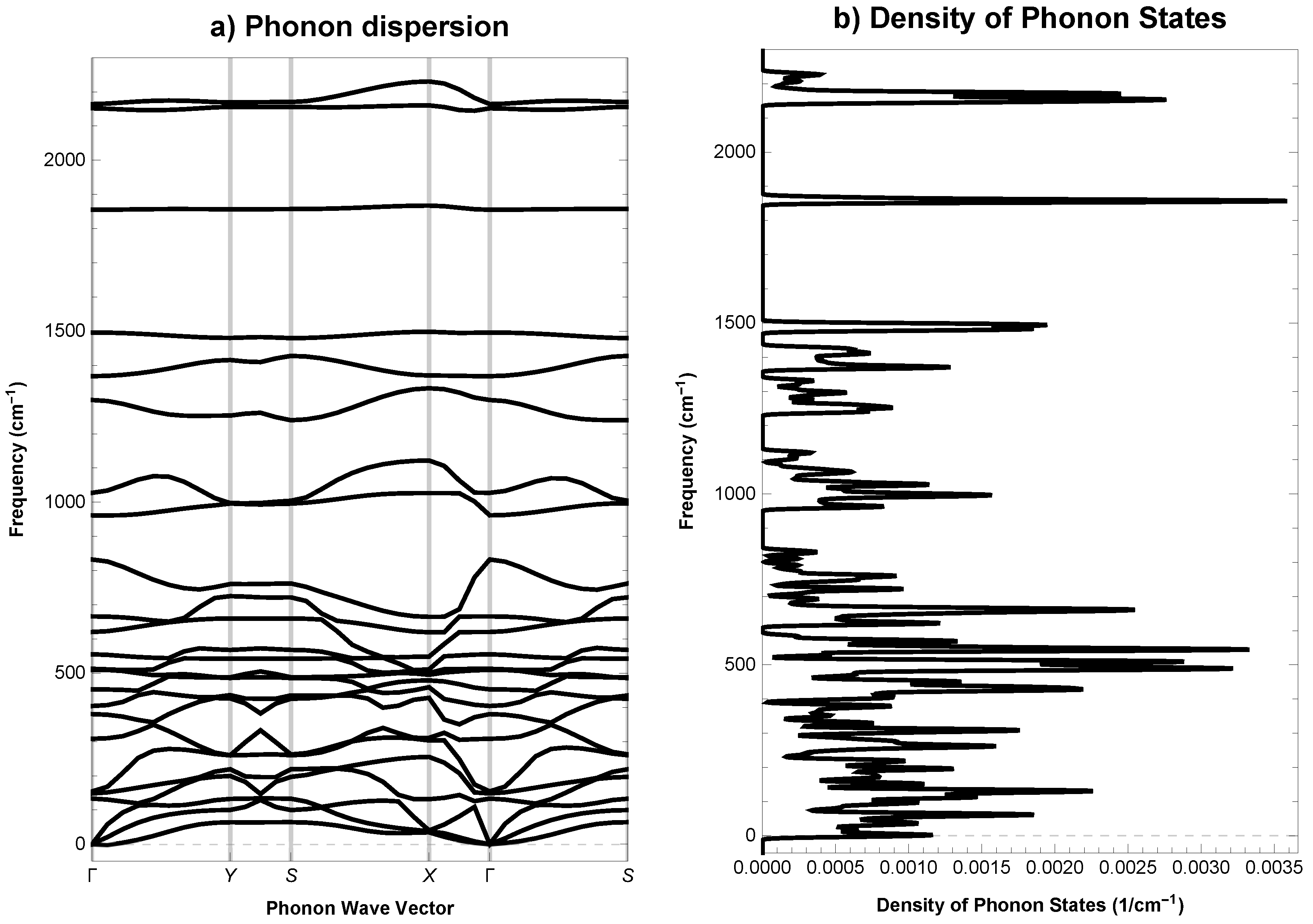
2.5. Mechanical and Phonon Properties
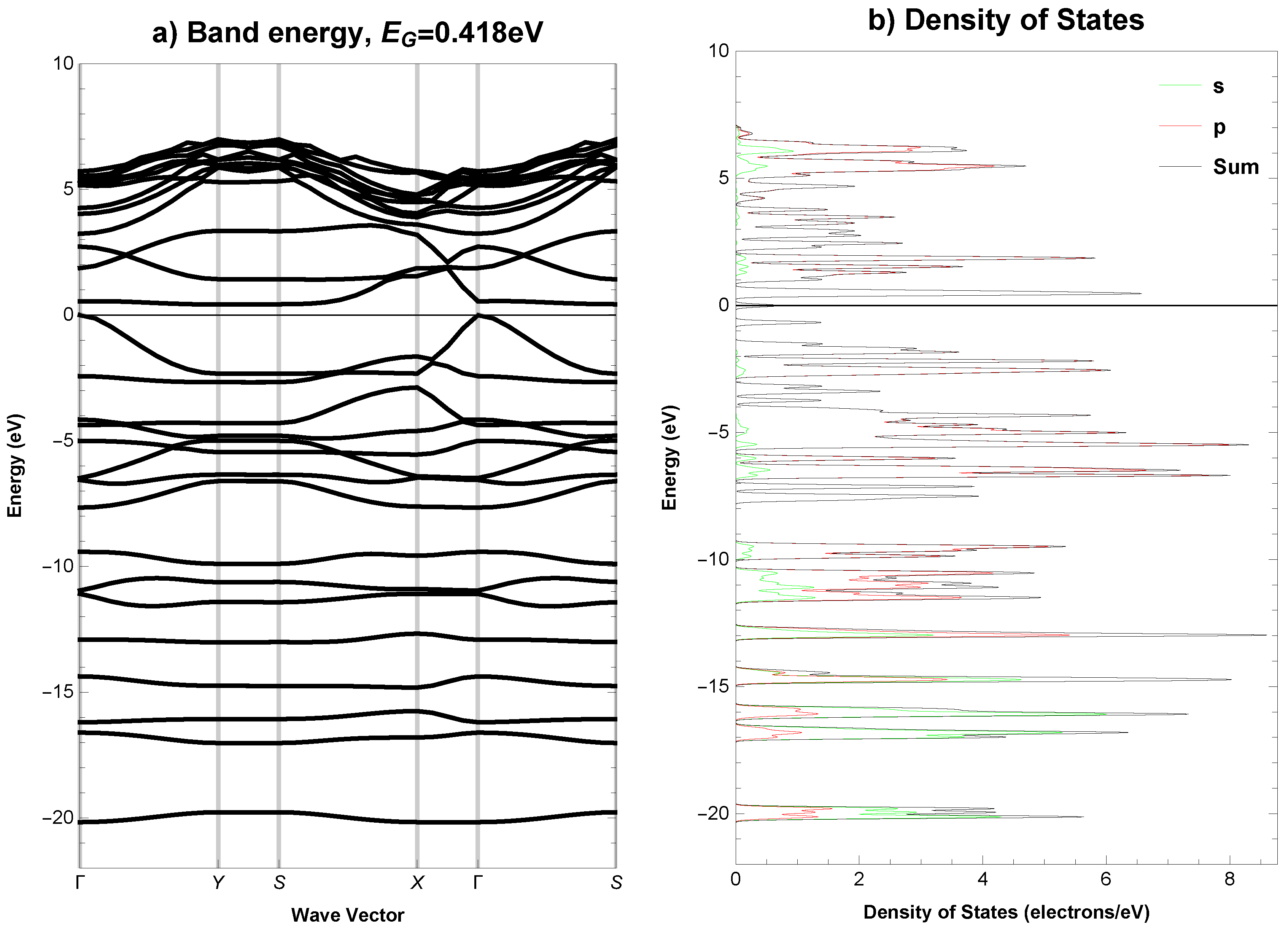
2.6. Electronic Properties
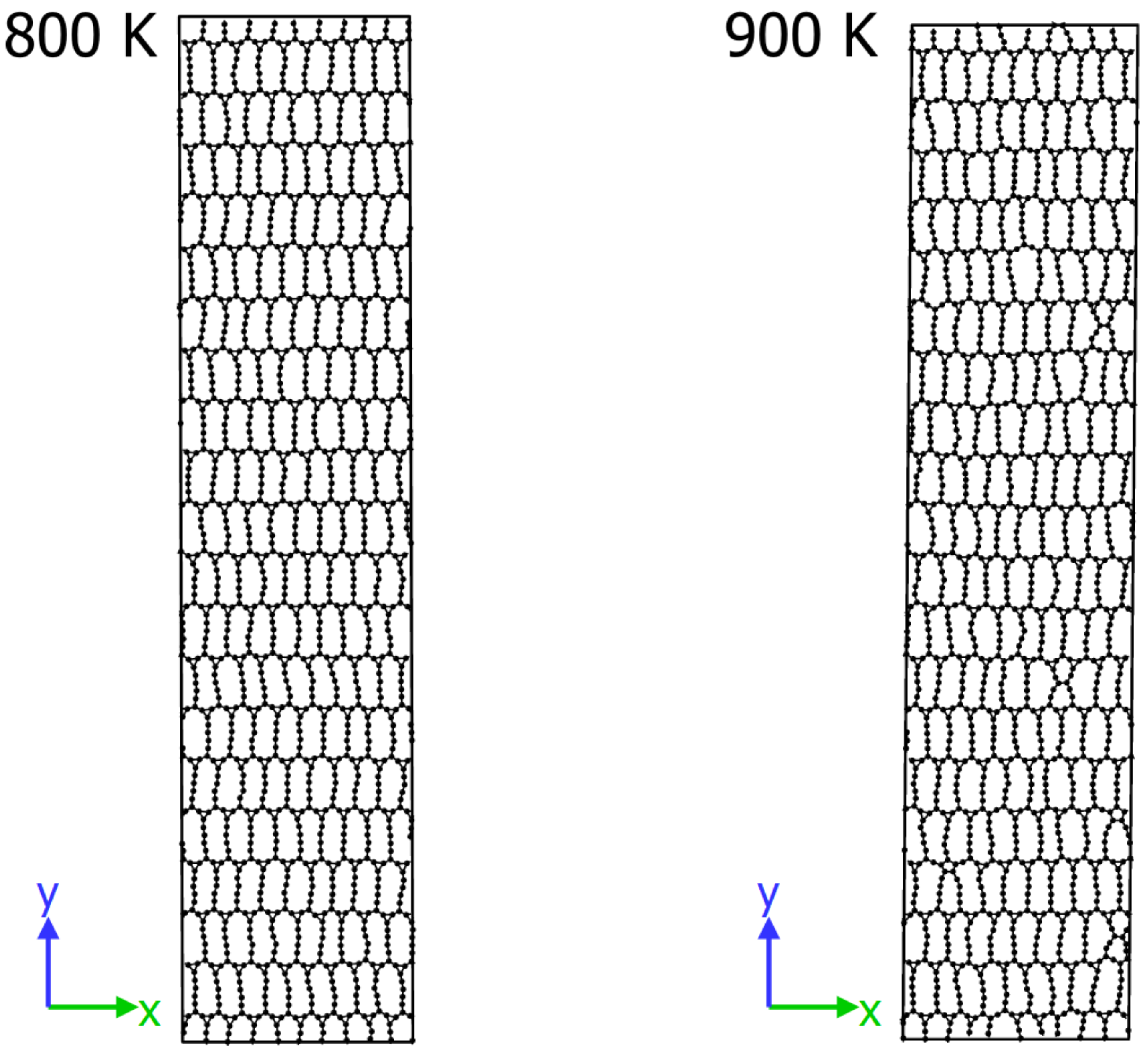
2.7. Finite Temperature Stability
3. Conclusions
- The proposed polymorph of graphene (rP16-P1m1) is mechanically and dynamically stable contrary to other C3-cyclicgraphenes.
- The proposed structure is thermally stable up to a temperature of at least 800 K.
- The relative energy of Anisotropic-cyclicgraphene with respect to pristine graphene is similar to other graphynes and cyclicgraphenes.
- Anisotropic-cyclicgraphene can be semiconducting, with a direct band gap with a value of 0.829 eV.
- The semi-empirical potential AIREBO seems to be surprisingly suitable for carbon structures.
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A. Unit Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lattice Parameters | ||
|---|---|---|
| a | 3.822 | |
| b | 16.967 | |
| Atom Number | Fractional coordinates of atoms | |
| 1 | 0.50000 | 0.28603 |
| 2 | 0.50000 | 0.36811 |
| 3 | 0.50000 | 0.44107 |
| 4 | 0.00000 | 0.78603 |
| 5 | 0.00000 | 0.86811 |
| 6 | 0.00000 | 0.94107 |
| 7 | 0.18150 | 0.24436 |
| 8 | 0.68150 | 0.74436 |
| 9 | 0.81850 | 0.24436 |
| 10 | 0.31850 | 0.74436 |
| 11 | 0.00000 | 0.17228 |
| 12 | 0.00000 | 0.01941 |
| 13 | 0.00000 | 0.09259 |
| 14 | 0.50000 | 0.67228 |
| 15 | 0.50000 | 0.51941 |
| 16 | 0.50000 | 0.59259 |
| Lattice Parameters | ||
|---|---|---|
| a | 3.822 | |
| b | 8.701 | |
| 77.1665° | ||
| Atom Number | Fractional coordinates of atoms | |
| 1 | 0.13221 | 0.59914 |
| 2 | 0.85619 | 0.51591 |
| 3 | 0.49331 | 0.51566 |
| 4 | 0.05066 | 0.76329 |
| 5 | 0.97724 | 0.90920 |
| 6 | 0.74632 | 0.37162 |
| 7 | 0.89892 | 0.06589 |
| 8 | 0.82547 | 0.21225 |
| Lattice Parameters | ||
|---|---|---|
| a | 3.922 | |
| b | 8.472 | |
| 90.0° | ||
| Atom Number | Fractional coordinates of atoms | |
| 1 | 0.15902 | 0.60266 |
| 2 | 0.85954 | 0.52400 |
| 3 | 0.45448 | 0.52020 |
| 4 | 0.14158 | 0.76639 |
| 5 | 0.02079 | 0.91373 |
| 6 | 0.67188 | 0.35866 |
| 7 | 0.89386 | 0.05964 |
| 8 | 0.77916 | 0.20769 |
References
- Heimann, R.; Evsvukov, S.; Koga, Y. Carbon allotropes: A suggested classification scheme based on valence orbital hybridization. Carbon 1997, 35, 1654–1658. [Google Scholar] [CrossRef]
- Hoffmann, R.; Kabanov, A.A.; Golov, A.A.; Proserpio, D.M. Homo Citans and Carbon Allotropes: For an Ethics of Citation. Angew. Chem. Int. Ed. 2016, 55, 10962–10976. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.; Novoselov, K. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.C.; Wang, R.Z.; Miao, M.S.; Wei, X.L.; Chen, Y.P.; Yan, H.; Lau, W.M.; Liu, L.M.; Ma, Y.M. Two dimensional Dirac carbon allotropes from graphene. Nanoscale 2014, 6, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Enyashin, A.N.; Ivanovskii, A.L. Graphene allotropes. Phys. Status Solidi (b) 2011, 248, 1879–1883. [Google Scholar] [CrossRef]
- Ivanovskii, A.L. Graphene-based and graphene-like materials. Russ. Chem. Rev. 2012, 81, 571–605. [Google Scholar] [CrossRef]
- Lu, X.; Chen, Z. Curved Pi-Conjugation, Aromaticity, and the Related Chemistry of Small Fullerenes (<C60) and Single-Walled Carbon Nanotubes. Chem. Rev. 2005, 105, 3643–3696. [Google Scholar] [PubMed]
- Appelhans, D.J.; Lin, Z.; Lusk, M.T. Two-dimensional carbon semiconductor: Density functional theory calculations. Phys. Rev. B 2010, 82, 073410. [Google Scholar] [CrossRef]
- Tan, C.; Cao, X.; Wu, X.J.; He, Q.; Yang, J.; Zhang, X.; Chen, J.; Zhao, W.; Han, S.; Nam, G.H.; et al. Recent Advances in Ultrathin Two-Dimensional Nanomaterials. Chem. Rev. 2017, 117, 6225–6331. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.H.; Ghosh, S.; Pradhan, B.; Dalui, A.; Shrestha, L.K.; Acharya, S.; Ariga, K. Two-Dimensional (2D) Nanomaterials towards Electrochemical Nanoarchitectonics in Energy-Related Applications. Bull. Chem. Soc. Jpn. 2017, 90, 627–648. [Google Scholar] [CrossRef]
- Fu, Q.; Bao, X. Surface chemistry and catalysis confined under two-dimensional materials. Chem. Soc. Rev. 2017, 46, 1842–1874. [Google Scholar] [CrossRef] [PubMed]
- Nasir, S.; Hussein, M.; Zainal, Z.; Yusof, N. Carbon-Based Nanomaterials/Allotropes: A Glimpse of Their Synthesis, Properties and Some Applications. Materials 2018, 11, 295. [Google Scholar] [CrossRef] [PubMed]
- Ivanovskii, A. Graphynes and graphdyines. Prog. Solid State Chem. 2013, 41, 1–19. [Google Scholar] [CrossRef]
- Wang, J.; Deng, S.; Liu, Z.; Liu, Z. The rare two-dimensional materials with Dirac cones. Natl. Sci. Rev. 2015, 2, 22–39. [Google Scholar] [CrossRef]
- Baughman, R.H.; Eckhardt, H.; Kertesz, M. Structure-property predictions for new planar forms of carbon: Layered phases containing sp2 and sp atoms. J. Chem. Phys. 1987, 87, 6687–6699. [Google Scholar] [CrossRef]
- Narita, N.; Nagai, S.; Suzuki, S.; Nakao, K. Optimized geometries and electronic structures of graphyne and its family. Phys. Rev. B 1998, 58, 11009–11014. [Google Scholar] [CrossRef]
- Li, G.; Li, Y.; Liu, H.; Guo, Y.; Li, Y.; Zhu, D. Architecture of graphdiyne nanoscale films. Chem. Commun. 2010, 46, 3256–3258. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Zuo, Z.; Yi, Y.; Liu, H.; Li, D.; Li, Y.; Li, Y. Low temperature, atmospheric pressure for synthesis of a new carbon Ene-yne and application in Li storage. Nano Energy 2017, 33, 343–349. [Google Scholar] [CrossRef]
- Ma, S.; Sun, L.Z.; Zhang, K.W. Prediction of two planar carbon allotropes with large meshes. Phys. Chem. Chem. Phys. 2016, 18, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Rao Nulakani, N.V.; Kamaraj, M.; Subramanian, V. Coro-graphene and circumcoro-graphyne: novel two-dimensional materials with exciting electronic properties. RSC Adv. 2015, 5, 78910–78916. [Google Scholar] [CrossRef]
- Song, Q.; Wang, B.; Deng, K.; Feng, X.; Wagner, M.; Gale, J.D.; Mullen, K.; Zhi, L. Graphenylene, a unique two-dimensional carbon network with nondelocalized cyclohexatriene units. J. Mater. Chem. C 2013, 1, 38–41. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, G.; Huang, Q.; Guo, L.; Chen, X. Structural and Electronic Properties of T Graphene: A Two-Dimensional Carbon Allotrope with Tetrarings. Phys. Rev. Lett. 2012, 108, 225505. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Li, H.D.; Wang, J.T. Structural stabilities and electronic properties of planar C4 carbon sheet and nanoribbons. Phys. Chem. Chem. Phys. 2012, 14, 11107–11111. [Google Scholar] [CrossRef] [PubMed]
- Nisar, J.; Jiang, X.; Pathak, B.; Zhao, J.; Kang, T.W.; Ahuja, R. Semiconducting allotrope of graphene. Nanotechnology 2012, 23, 385704. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Liu, L.M.; Hu, Z.; Wang, W.H.; Song, W.X.; Li, F.; Zhao, S.J.; Liu, H.; Wang, H.T.; Tian, Y. Two-Dimensional Superlattice: Modulation of Band Gaps in Graphene-Based Monolayer Carbon Superlattices. J. Phys. Chem. Lett. 2012, 3, 3373–3378. [Google Scholar] [CrossRef]
- Jiang, J.W.; Leng, J.; Li, J.; Guo, Z.; Chang, T.; Guo, X.; Zhang, T. Twin graphene: A novel two-dimensional semiconducting carbon allotrope. Carbon 2017, 118, 370–375. [Google Scholar] [CrossRef]
- Hu, M.; Shu, Y.; Cui, L.; Xu, B.; Yu, D.; He, J. Theoretical two-atom thick semiconducting carbon sheet. Phys. Chem. Chem. Phys. 2014, 16, 18118–18123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, J.; Wang, Q.; Chen, X.; Kawazoe, Y.; Jena, P. Penta-graphene: A new carbon allotrope. Proc. Natl. Acad. Sci. USA 2015, 112, 2372–2377. [Google Scholar] [CrossRef] [PubMed]
- Einollahzadeh, H.; Dariani, R.; Fazeli, S. Computing the band structure and energy gap of penta-graphene by using DFT and G0W0 approximations. Solid State Commun. 2016, 229, 1–4. [Google Scholar] [CrossRef]
- Ewels, C.P.; Rocquefelte, X.; Kroto, H.W.; Rayson, M.J.; Briddon, P.R.; Heggie, M.I. Predicting experimentally stable allotropes: Instability of penta-graphene. Proc. Natl. Acad. Sci. USA 2015, 112, 15609–15612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrozek, A.; Kuś, W.; Burczyński, T. Nano level optimization of graphene allotropes by means of a hybrid parallel evolutionary algorithm. Comput. Mater. Sci. 2015, 106, 161–169. [Google Scholar] [CrossRef]
- Kuś, W.; Mrozek, A.; Burczyński, T. Memetic Optimization of Graphene-Like Materials on Intel PHI Coprocessor. In Proceedings of the Artificial Intelligence and Soft Computing: 15th International Conference (ICAISC 2016), Zakopane, Poland, 12–16 June 2016; Part I. Springer International Publishing: Cham, Switzerland, 2016; pp. 401–410. [Google Scholar]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Tadmor, E.B.; Miller, R.E. Modeling Materials: Continuum, Atomistic and Multiscale Techniques; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar]
- Maździarz, M.; Young, T.D.; Jurczak, G. A study of the affect of prerelaxation on the nanoindentation process of crystalline copper. Arch. Mech. 2011, 63, 533–548. [Google Scholar]
- Maździarz, M.; Young, T.D.; Dłuzewski, P.; Wejrzanowski, T.; Kurzydłowski, K.J. Computer modelling of nanoindentation in the limits of a coupled molecular–statics and elastic scheme. J. Comput. Theor. Nanosci. 2010, 7, 1172–1181. [Google Scholar] [CrossRef]
- Mrozek, A.; Kuś, W.; Burczyński, T. Method for determining structures on new carbon-based 2D materials with predefined mechanical properties. Int. J. Multiscale Comput. Eng. 2017, 15, 379–394. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M. First principles methods using CASTEP. Z. Kristall. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed]
- CASTEP GUIDE; BIOVIA: San Diego, CA, USA, 2014; Available online: http://www.tcm.phy.cam.ac.uk/castep/documentation/WebHelp/content/pdfs/castep.htm (accessed on 1 March 2018).
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef] [PubMed]
- Maździarz, M.; Mrozek, A.; Kuś, W.; Burczyński, T. First-principles study of new X-graphene and Y-graphene polymorphs generated by the two stage strategy. Mater. Chem. Phys. 2017, 202, 7–14. [Google Scholar] [CrossRef]
- Grabowski, B.; Ismer, L.; Hickel, T.; Neugebauer, J. Ab initio up to the melting point: Anharmonicity and vacancies in aluminum. Phys. Rev. B 2009, 79, 134106. [Google Scholar] [CrossRef]
- Cranford, S.W.; Buehler, M.J. Mechanical properties of graphyne. Carbon 2011, 49, 4111–4121. [Google Scholar] [CrossRef]
- Stuart, S.J.; Tutein, A.B.; Harrison, J.A. A reactive potential for hydrocarbons with intermolecular interactions. J. Chem. Phys. 2000, 112, 6472–6486. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO—The Open Visualization Tool. Modelling Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Long, G.; Zhou, Y.; Jin, M.; Kan, B.; Zhao, Y.; Gray-Weale, A.; Jiang, D.; Chen, Y.; Zhang, Q. Theoretical investigation on two-dimensional non-traditional carbon materials employing three-membered ring and four-membered ring as building blocks. Carbon 2015, 95, 1033–1038. [Google Scholar] [CrossRef]
- Zhao, J.; Wei, N.; Fan, Z.; Jiang, J.W.; Rabczuk, T. The mechanical properties of three types of carbon allotropes. Nanotechnology 2013, 24, 095702. [Google Scholar] [CrossRef] [PubMed]
- Fox, M.; Whitesell, J. Organic Chemistry; Jones and Bartlett Publishers: Burlington, MA, USA, 2004. [Google Scholar]
- Hou, X.; Xie, Z.; Li, C.; Li, G.; Chen, Z. Study of Electronic Structure, Thermal Conductivity, Elastic and Optical Properties of α, β, γ-Graphyne. Materials 2018, 11, 188. [Google Scholar]
- Andrew, R.C.; Mapasha, R.E.; Ukpong, A.M.; Chetty, N. Mechanical properties of graphene and boronitrene. Phys. Rev. B 2012, 85, 125428. [Google Scholar] [CrossRef]
- Yue, Q.; Chang, S.; Kang, J.; Qin, S.; Li, J. Mechanical and Electronic Properties of Graphyne and Its Family under Elastic Strain: Theoretical Predictions. J. Phys. Chem. C 2013, 117, 14804–14811. [Google Scholar] [CrossRef]







| Source | DFT | MD |
|---|---|---|
| a | 3.822 (3.822) | 3.936 (3.936) |
| b | 16.967 (8.701) | 17.544 (9.000) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maździarz, M.; Mrozek, A.; Kuś, W.; Burczyński, T. Anisotropic-Cyclicgraphene: A New Two-Dimensional Semiconducting Carbon Allotrope. Materials 2018, 11, 432. https://doi.org/10.3390/ma11030432
Maździarz M, Mrozek A, Kuś W, Burczyński T. Anisotropic-Cyclicgraphene: A New Two-Dimensional Semiconducting Carbon Allotrope. Materials. 2018; 11(3):432. https://doi.org/10.3390/ma11030432
Chicago/Turabian StyleMaździarz, Marcin, Adam Mrozek, Wacław Kuś, and Tadeusz Burczyński. 2018. "Anisotropic-Cyclicgraphene: A New Two-Dimensional Semiconducting Carbon Allotrope" Materials 11, no. 3: 432. https://doi.org/10.3390/ma11030432