Direct Utilization of Liquid Fuels in SOFC for Portable Applications: Challenges for the Selection of Alternative Anodes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: Principles, Typical Materials and Applications

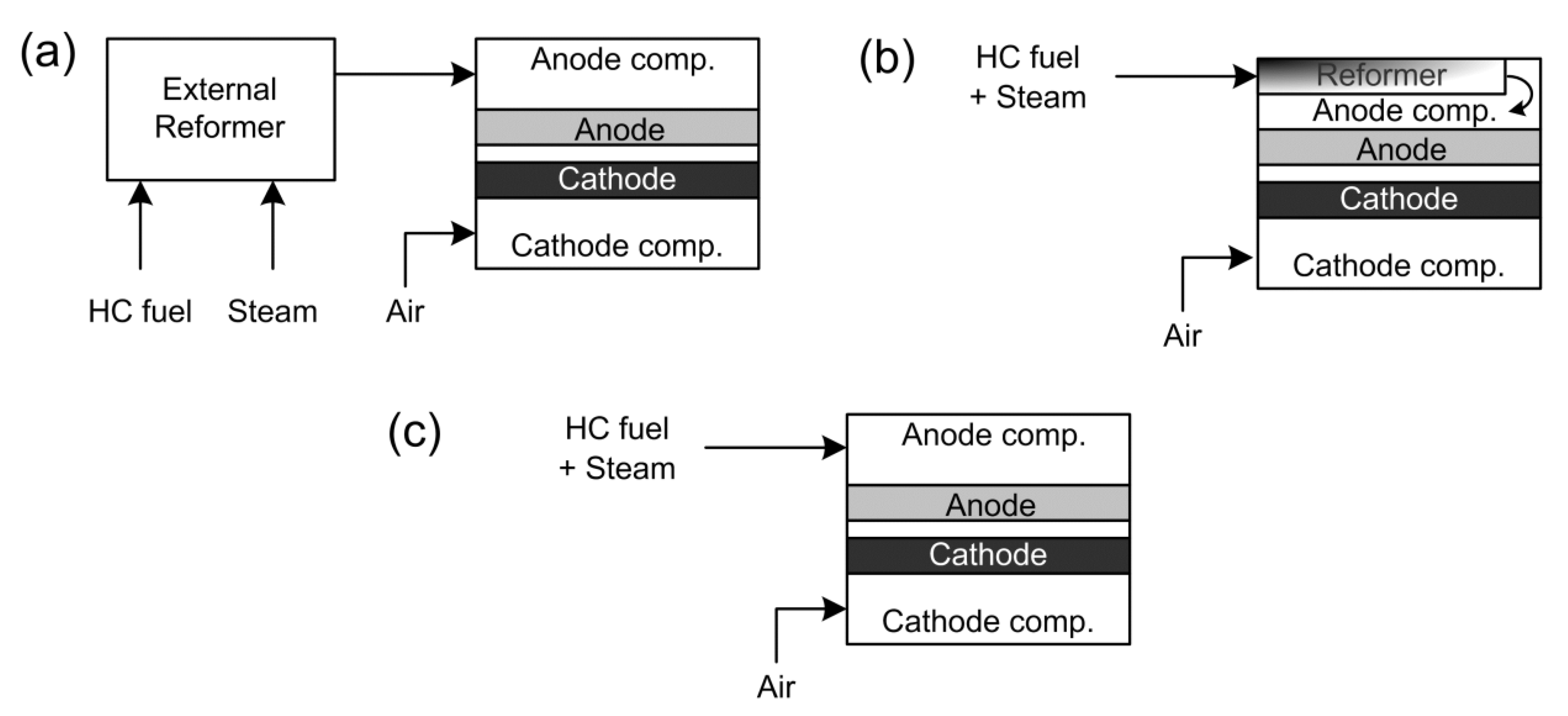
2. SOFC Operating Modes with Hydrocarbon Fuels

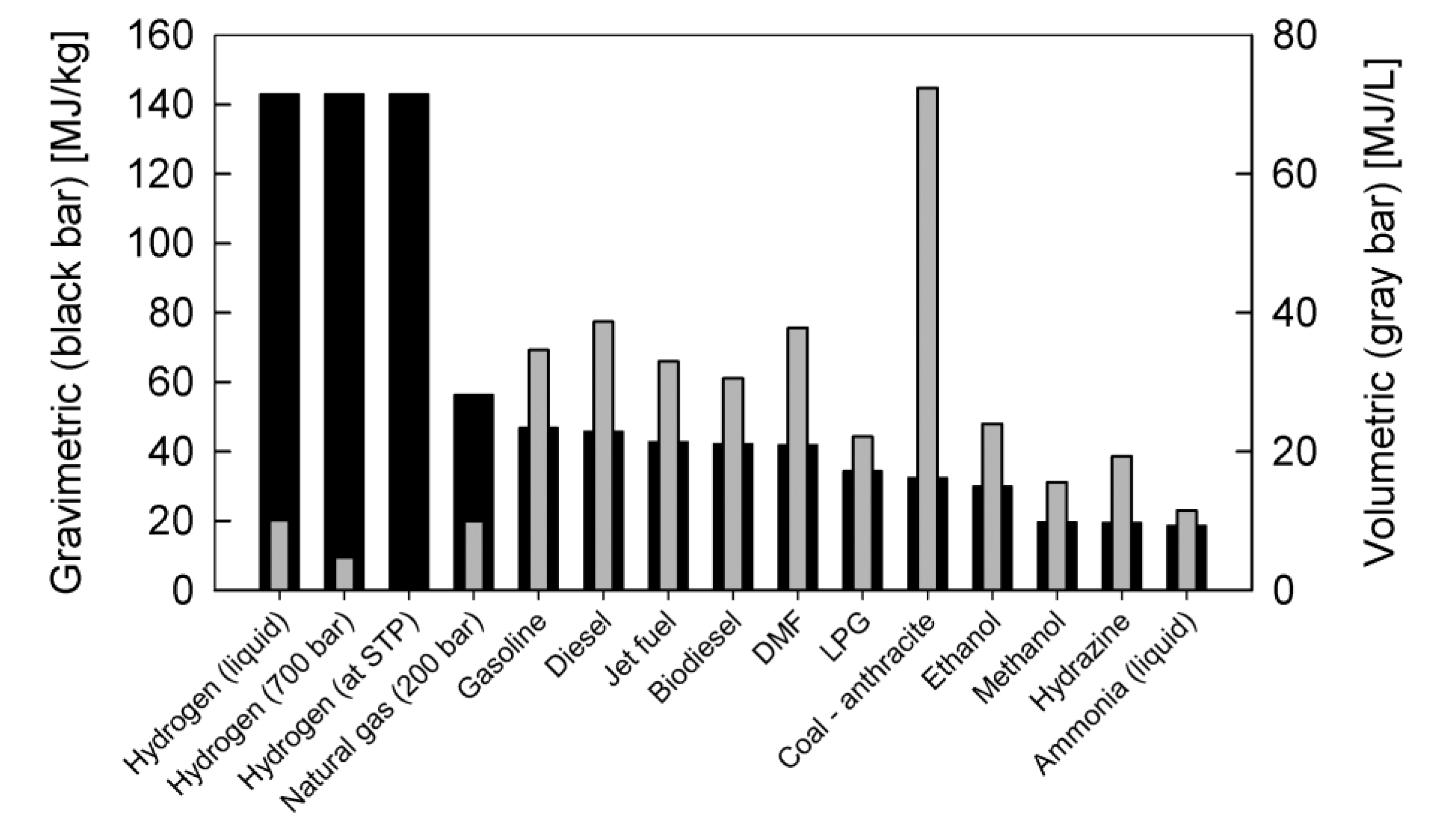
3. Liquid Fuels for SOFC
3.1. Methanol

3.2. Ethanol
3.3. Dimethyl Ether, Liquefied Petroleum Gas, and Other Conventional Liquid Fuels
3.4. Carbon-free Liquid Fuels
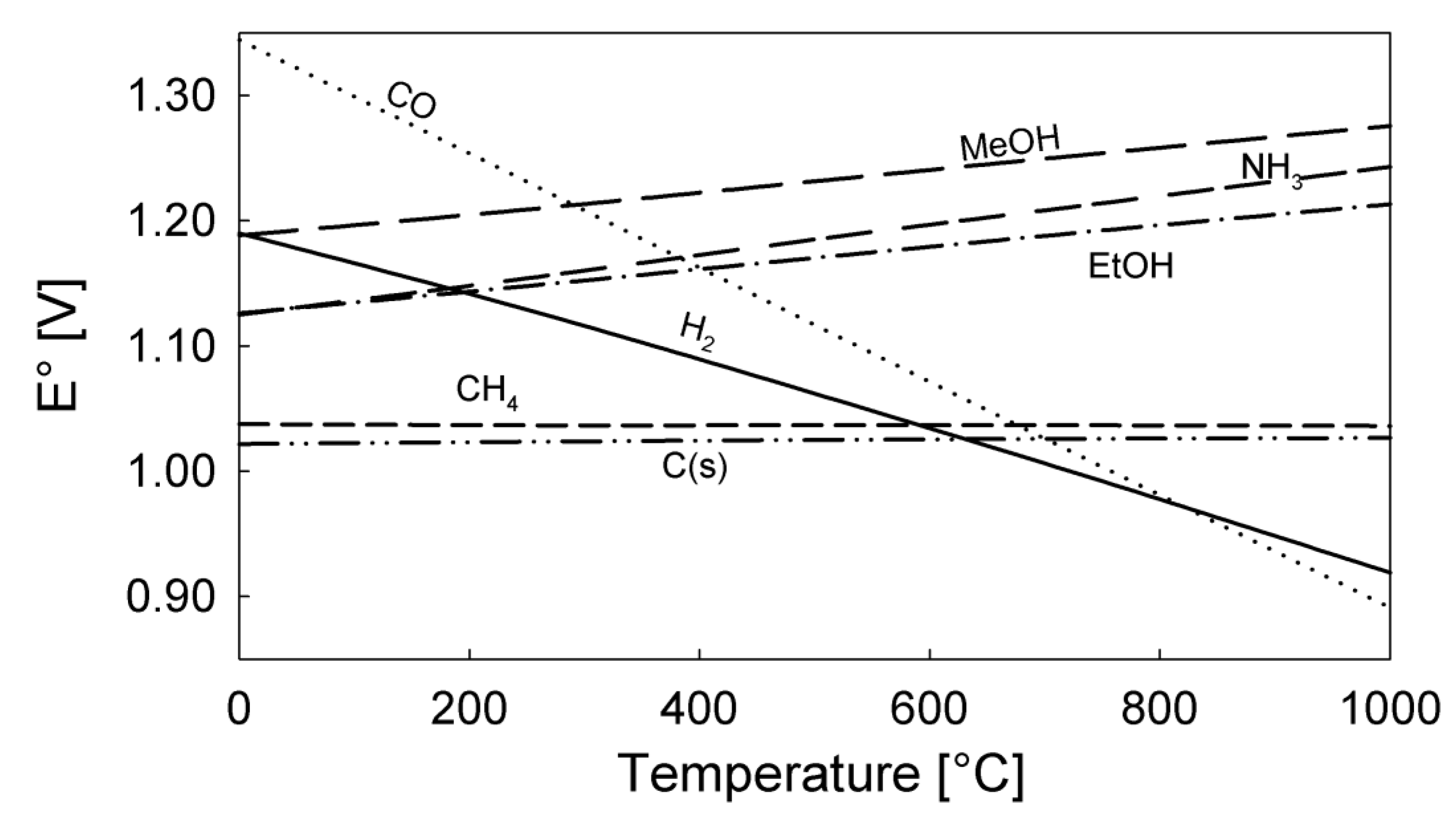
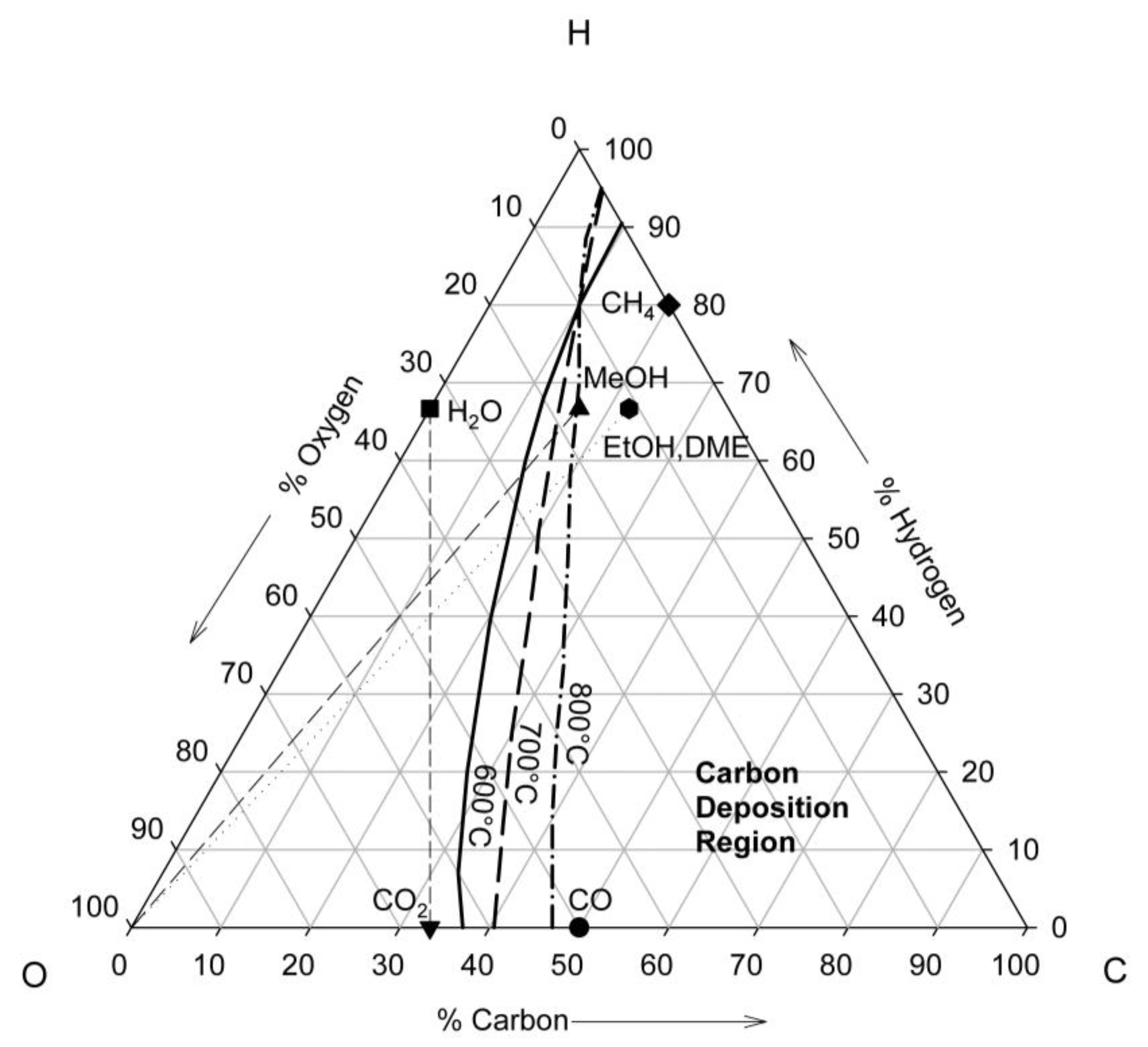
4. Thermodynamics of Direct Utilization SOFC




5. Kinetics in the Anode Compartment


6. Survey of Anode Materials for the Direct Utilization of Hydrocarbons and Liquid Fuels
6.1. Early Studies on Alternative Anodes
6.2. Metal/Ceria Anodes
6.3. Mixed-conductor Oxides Anodes
7. Direct Utilization of MeOH and EtOH

8. Way Forward
- The anode is the site of electrocatalytic oxidation; therefore, its composition should provide reactivity and electron conductivity simultaneously.
- The anode should withstand the typical SOFC operating conditions (i.e. high temperature T = 600–1,000 ºC, and highly reducing environment pO2 = 10-20 atm).
- The anode should be compatible with the electrolyte and with the current collector materials (i.e. matching thermal expansion and possessing mutual chemical inertia).
- The anode should be stable and maintain its structural integrity after repeated temperature and redox cycling (chemical atmosphere).
- The anode should be inert to fuels as well as to the oxidation products (i.e. it should not react chemically with fuels or oxidants).
- The materials should be inexpensive and the preparation process should be simple and cheap to manufacture.
- The anodic microstructure should tolerate and possibly inhibit the deposition of carbon and tars (i.e. coke deactivation should be reversible, so that once the carbon deposits are removed the anode should regain the initial morphology and performance).
- The anode should be able to oxidize the main species composing the fuel, particularly for those fuels that are readily decomposed by pyrolysis of catalytic decomposition, and should be selective for the complete oxidation.
Acknowledgements
References and Notes
- Singhal, S.C.; Kendall, K.E. High Temperature Solid Oxide Fuel Cells - Fundamentals, Design and Applications, 1st ed.; Elsevier: Oxford, U.K, 2003; p. 405. [Google Scholar]
- Minh, N.Q.; Takahashi, T. Science and Technology of Ceramic Fuel Cells; Elsevier: Amsterdam, The Netherlands, 1995; p. 366. [Google Scholar]
- Maguire, E.; Gharbage, B.; Marques, F.M.B.; Labrincha, J.A. Cathode Materials for Intermediate Temperature SOFCs. Solid State Ionics 2000, 127, 329–335. [Google Scholar] [CrossRef]
- Lee, S.; Lim, Y.; Lee, E.A.; Hwang, H.J.; Moon, J.-W. Ba0.5Sr0.5Co0.8Fe0.2O3-δ (BSCF) and La0.6Ba0.4Co0.2Fe0.8O3-δ (LBCF) Cathodes Prepared by Combined Citrate-EDTA Method for IT-SOFCs. J. Power Sources 2006, 157, 848–854. [Google Scholar]
- Baumann, F.S.; Fleig, J.; Habermeier, H.U.; Maier, J. Impedance Spectroscopic Study on Well-Defined (La,Sr)(Co,Fe)O3-d Model Electrodes. Solid State Ionics 2006, 177, 1071–1081. [Google Scholar] [CrossRef]
- Baumann, F.S.; Fleig, J.; Habermeier, H.U.; Maier, J. Ba0.5Sr0.5Co0.8Fe0.2O3-δ Thin Film Microelectrodes Investigated by Impedance Spectroscopy. Solid State Ionics 2006, 177, 3187–3191. [Google Scholar]
- Scott, S.P.; Mantzavinos, D.; Hartley, A.; Sahibzada, M.; Metcalfe, I.S. Reactivity of LSCF Perovskites. Solid State Ionics 2002, 152, 777–781. [Google Scholar] [CrossRef]
- Liu, J.; Barnett, S.A. Thin Yttrium-Stabilized Zirconia Electrolyte Solid Oxide Fuel Cells by Centrifugal Casting. J. Amer. Ceram. Soc. 2002, 85, 3096–3098. [Google Scholar] [CrossRef]
- Fuel Cell Handbook, 7th ed.U.S. Department of Energy, Office of Fossil Energy, National Energy Technology Laboratory: Morgantown, WV, USA, 2004.
- Vielstich, W.; Lamm, A.; Gasteiger, H.E. Handbook of Fuel Cells - Fundamentals Technology and Applications, 1st ed.; John Wiley & Sons Ltd: West Sussex, U.K, 2003; p. 439. [Google Scholar]
- De Souza, S.; Visco, S.J.; De Jonghe, L.C. Thin-Film Solid Oxide Fuel Cell with High Performance at Low-Temperature. Solid State Ionics 1997, 98, 57–61. [Google Scholar] [CrossRef]
- Kim, J.-W.; Virkar, A.V.; Fung, K.-Z.; Mehta, K.; Singhal, S.C. Polarization Effects in Intermediate Temperature, Anode-Supported Solid Oxide Fuel Cells. J. Electrochem. Soc. 1999, 146, 69–78. [Google Scholar] [CrossRef]
- Matsuzaki, Y.; Yasuda, I. The Poisoning Effect of Sulfur-Containing Impurity Gas on a SOFC Anode: Part I. Dependence on Temperature, Time, and Impurity Concentration. Solid State Ionics 2000, 132, 261–269. [Google Scholar] [CrossRef]
- Sarantaridis, D.; Atkinson, A. Redox Cycling of Ni-Based Solid Oxide Fuel Cell Anodes: A Review. Fuel Cells 2007, 7, 246–258. [Google Scholar] [CrossRef]
- Singhal, S.C. Solid Oxide Fuel Cells for Stationary, Mobile, and Military Applications. Solid State Ionics 2002, 152, 405–410. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R. Industrial Catalysis, the Science and the Challenge: Conversion of Fossil Fuels. Catal. Today 1993, 18, 125–145. [Google Scholar] [CrossRef]
- Peters, R.; Dahl, R.; Kluttgen, U.; Palm, C.; Stolten, D. Internal Reforming of Methane in Solid Oxide Fuel Cell Systems. J. Power Sources 2002, 106, 238–244. [Google Scholar] [CrossRef]
- Ormerod, R.M. Internal reforming in solid oxide fuel cells. In Proceedings of the International Symposium on Reaction Kinetics and the Development of Catalytic Processes, Brugge, Belgium, April 1999; pp. 35–46.
- Liu, J.A.; Barnett, S.A. Operation of Anode-Supported Solid Oxide Fuel Cells on Methane and Natural Gas. Solid State Ionics 2003, 158, 11–16. [Google Scholar] [CrossRef]
- Lee, A.L.; Zabransky, R.F.; Huber, W.J. Internal Reforming Development for Solid Oxide Fuel Cells. Ind. Eng. Chem. Res. 1990, 29, 766–773. [Google Scholar] [CrossRef]
- Finnerty, C.M.; Ormerod, R.M. Internal Reforming Over Nickel/Zirconia Anodes in SOFCS Oparating on Methane: Influence of Anode Formulation, Pre-Treatment and Operating Conditions. J. Power Sources 2000, 86, 390–394. [Google Scholar] [CrossRef]
- Klein, J.M.; Henault, M.; Gelin, P.; Bultel, Y.; Georges, S. A Solid Oxide Fuel Cell Operating in Gradual Internal Reforming Conditions under Pure Dry Methane. Electrochem. Solid State Lett. 2008, 11, B144–B147. [Google Scholar] [CrossRef]
- Cheekatamarla, P.K.; Finnerty, C.M.; Cai, J. Internal Reforming of Hydrocarbon Fuels in Tubular Solid Oxide Fuel Cells. Int. J. Hydrogen. Energ. 2008, 33, 1853–1858. [Google Scholar] [CrossRef]
- Zhan, Z.; Barnett, S.A. An Octane-Fueled Solid Oxide Fuel Cell. Science 2005, 308, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.T.; Chadwick, D.; Kershenbaum, L. Achieving Autothermal Operation in Internally Reformed Solid Oxide Fuel Cells: Experimental Studies. Ind. Eng. Chem. Res. 2007, 46, 8518–8524. [Google Scholar] [CrossRef]
- Janardhanan, V.M.; Heuveline, V.; Deutschmann, O. Performance Analysis of a SOFC under Direct Internal Reforming Conditions. J. Power Sources 2007, 172, 296–307. [Google Scholar] [CrossRef]
- Zhan, Z.; Barnett, S.A. Use of a Catalyst Layer for Propane Partial Oxidation in Solid Oxide Fuel Cells. Solid State Ionics 2005, 176, 871–879. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, W.; Ran, R.; Cai, R.; Shao, Z.P. Methane-Fueled SOFC with Traditional Nickel-Based Anode by Applying Ni/Al2O3 as a Dual-Functional Layer. Electrochem. Commun. 2009, 11, 194–197. [Google Scholar] [CrossRef]
- Pillai, M.R.; Bierschenk, D.M.; Barnett, S.A. Electrochemical Partial Oxidation of Methane in Solid Oxide Fuel Cells: Effect of Anode Reforming Activity. Catalysis Letter. 2008, 121, 19–23. [Google Scholar] [CrossRef]
- Bockris, J.O.M.; Reddy, A.K.N.; Gamboa-Aldeco, M. Modern Electrochemistry, 2nd ed.; Kluwer Academic/Plenum Publisher: New York, NY, USA, 2000; p. 1534. [Google Scholar]
- Gorte, R.J.; Kim, H.; Vohs, J.M. Novel SOFC Anodes for the Direct Electrochemical Oxidation of Hydrocarbon. J. Power Sources 2002, 106, 10–15. [Google Scholar] [CrossRef]
- Mogensen, M.; Kammer, K. Conversion of Hydrocarbons in Solid Oxide Fuel Cells. Ann. Rev. Mater. Res. 2003, 33, 321–331. [Google Scholar] [CrossRef]
- Gorte, R.J.; Park, S.; Vohs, J.M.; Wang, C.H. Anodes for Direct Oxidation of Dry Hydrocarbons in a Solid-Oxide Fuel Cell. Adv. Mater. 2000, 12, 1465–1469. [Google Scholar] [CrossRef]
- Gorte, R.J.; Vohs, J.M. Novel SOFC Anodes for the Direct Electrochemical Oxidation of Hydrocarbons. J. Catal. 2003, 216, 477–486. [Google Scholar] [CrossRef]
- Kim, H.; Park, S.; Vohs, J.M.; Gorte, R.J. Direct Oxidation of Liquid Fuels in a Solid Oxide Fuel Cell. J. Electrochem. Soc. 2001, 148, A693–A695. [Google Scholar] [CrossRef]
- Horita, T.; Yamaji, K.; Kato, T.; Kishimoto, H.; Xiong, Y.; Sakai, N.; Brito, M.E.; Yokokawa, H. Imaging of CH4 Decomposition Around the Ni/YSZ Interfaces under Anodic Polarization. J. Power Sources 2005, 145, 133–138. [Google Scholar] [CrossRef]
- Horita, T.; Yamaji, K.; Kato, T.; Sakai, N.; Yokokawa, H. Comparison of Catalytic Activity for CH4 Decomposition at the Metal/Oxide Interfaces by Isotope-Labeling Technique. Solid State Ionics 2004, 172, 93–99. [Google Scholar] [CrossRef]
- Horita, T.; Yamaji, K.; Kato, T.; Sakai, N.; Yokokawa, H. Design of Metal/Oxide Interfaces for the Direct Introduction of Hydrocarbons into SOFCs. J. Power Sources 2004, 131, 299–303. [Google Scholar] [CrossRef]
- Cheng, W.H.; Kung, H.H. Methanol Production and Use; Marcel Dekker, Inc.: New York, NY, USA, 1994; p. 326. [Google Scholar]
- The Bureau of Mines Synthetic Liquid Fuels Program, 1944-55; Bureau of Mines Bituminous Coal Staff, US Dept of the Interior: Washington, D.C., USA., 1959.
- Olah, G.A. Beyond Oil and Gas: the Methanol Economy; Wiley-VCH Weinheim: Berlin, Germany, 2006; p. 290. [Google Scholar]
- Olah, G.A. The Methanol Economy. Chem. Eng. News 2003, 81, 5. [Google Scholar]
- Olah, G.A. After Oil and Gas: Methanol Economy. Catalysis Lett. 2004, 93, 1–2. [Google Scholar] [CrossRef]
- Sahibzada, M.; Metcalfe, I.S.; Chadwick, D. Methanol Synthesis from CO2/H2 Over Pd Promoted Cu/ZnO/Al2O3 Catalysts. In Advances in Chemical Conversions for Mitigating Carbon Dioxide, Proceedings of the Fourth International Conference on Carbon Dioxide Utilization, Kyoto, Japan, September 1997; pp. 351–356.
- Liebhafsky, H.A.; Cairns, E.J. Fuel Cells and Fuel Batteries - A Guide to Their Research and Development; John Wiley & Sons, Inc.: New York, NY, USA, 1968; p. 692. [Google Scholar]
- Yaws, C.L. The Yaws Handbook of Physical Properties for Hydrocarbons and Chemicals : Physical Properties for More Than 41,000 Organic and Inorganic Chemical Compounds : Coverage for C1 to C100 Organics and Ac to Zr Inorganics; Gulf Publishing Company: Houston, TX, USA, 2005; p. 812. [Google Scholar]
- Termite Power. Available online: http://www.jgi.doe.gov/education/bioenergy/ bioenergy_4.html (accessed on 24 December 2007).
- Austin, G.T. Shreve's Chemical Process Industries, 5th ed.; McGraw-Hill Book Company: New York, NY, USA, 1984; p. 859. [Google Scholar]
- Nichols, R.J. The Methanol Story: A Sustainable Fuel for the Future. J. Sci. Ind. Res. India. 2003, 62, 97–105. [Google Scholar]
- Nichols, R.J. The Challenges of Change in the Auto Industry - Why Alternative Fuels. J. Eng. Gas Turb. Power-T Asme. 1994, 116, 727–732. [Google Scholar] [CrossRef]
- Canadial Renewable Fuel Association. Available online: www.greenfuels.org/ (accessed on 2 December 2007).
- Bourne, J.K., Jr. Green Dreams: Making Fuel from Crops Could be Good for the Planet-After a Breakthrough or Two.(Cover Story). In National Geographic; National Geographic Society: New York, NY, USA, 2007; p. 38. [Google Scholar]
- Pearce, F.; Aldhous, P. Is the biofuel dream over? New Sci. 2007, 2634, 6–7. [Google Scholar]
- The Energy Balance of Corn Ethanol: An Update. Available online: http://www.transportation.anl.gov/pdfs/AF/265.pdf (accessed on August 2007).
- Vigier, F.; Coutanceau, C.; Perrard, A.; Belgsir, E.M.; Lamy, C. Development of Anode Catalysts for a Direct Ethanol Fuel Cell. J. Appl. Electrochem. 2004, 34, 439–446. [Google Scholar] [CrossRef]
- Ni, M.; Leung, D.Y.C.; Leung, M.K.H. A Review on Reforming Bio-Ethanol for Hydrogen Production. Int. J. Hydrogen. Energ. 2007, 32, 3238–3247. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R.; Nielsen, R. Fuels and Energy for the Future: The Role of Catalysis. Catal. Rev-Sci. Eng. 2004, 46, 247–270. [Google Scholar] [CrossRef]
- Haldor-Topsoe: Pioneering DME. Available online: http://www.topsoe.com/Business_areas/ Gasification-based/Processes/Dimethylether.aspx (accessed on February 2009).
- Moon, D.J. Hydrogen Production by Catalytic Reforming of Gaseous Hydrocarbons (Methane & LPG). Catalysis Surveys from Asia 2008, 12, 188–202. [Google Scholar]
- Chen, Y.Z.; Shao, Z.P.; Xu, N.P. Partial Oxidation of Dimethyl Ether to H2/Syngas over Supported Pt Catalyst. J. Nat. Gas Chem. 2008, 17, 75–80. [Google Scholar] [CrossRef]
- Laosiripojana, N.; Assabumrungrat, S. Catalytic Steam Reforming of Dimethyl Ether (DME) over High Surface Area Ce-ZrO2 at SOFC Temperature: The Possible Use of DME in Indirect Internal Reforming Operation (IIR-SOFC). Appl. Catal. A-Gen. 2007, 320, 105–113. [Google Scholar] [CrossRef]
- Murray, E.P.; Harris, S.J.; Jen, H.W. Solid Oxide Fuel Cells Utilizing Dimethyl Ether Fuel. J. Electrochem. Soc. 2002, 149, A1127–A1131. [Google Scholar] [CrossRef]
- Murray, E.P.; Harris, S.J.; Liu, J.; Barnett, S.A. Direct Solid Oxide Fuel Cell Operation Using a Dimethyl Ether/Air Fuel Mixture. Electrochem. Solid State Lett. 2005, 8, A531–A533. [Google Scholar] [CrossRef]
- Chen, F.Z.; Zha, S.W.; Dong, J.; Liu, M.L. Pre-reforming of Propane for Low-Temperature SOFCs. Solid State Ionics 2004, 166, 269–273. [Google Scholar] [CrossRef]
- Hibino, T.; Hashimoto, A.; Inoue, T.; Tokuno, J.; Yoshida, S.; Sano, M. A Low-Operating-Temperature Solid Oxide Fuel Cell in Hydrocarbon-Air Mixtures. Science 2000, 288, 2031–2033. [Google Scholar] [CrossRef] [PubMed]
- Rampe, T.; Heinzel, A.; Vogel, B. Hydrogen Generation from Biogenic and Fossil Fuels by Autothermal Reforming. J. Power Sources 2000, 86, 536–541. [Google Scholar] [CrossRef]
- Zhan, Z.; Liu, J.; Barnett, S.A. Operation of Anode-Supported Solid Oxide Fuel Cells on Propane-Air Fuel Mixtures. Appl. Catal. A-Gen. 2004, 262, 255–259. [Google Scholar] [CrossRef]
- Hibino, T.; Hashimoto, A.; Yano, M.; Suzuki, M.; Sano, M. Ru-Catalyzed Anode Materials for Direct Hydrocarbon SOFCs. Electrochim. Acta 2003, 48, 2531–2537. [Google Scholar] [CrossRef]
- Madsen, B.D.; Barnett, S.A. Effect of Fuel Composition on the Performance of Ceramic-Based Solid Oxide Fuel Cell Anodes. Solid State Ionics 2005, 176, 2545–2553. [Google Scholar] [CrossRef]
- Wojcik, A.; Middleton, H.; Damopoulos, I.; van Herle, J. Ammonia as a Fuel in Solid Oxide Fuel Cells. J. Power Sources 2003, 118, 342–348. [Google Scholar] [CrossRef]
- Zhang, L.M.; Cong, Y.; Yang, W.S.; Lin, L.W. A direct ammonia tubular solid oxide fuel cell. Chin. J. Catal. 2007, 28, 749–751. [Google Scholar] [CrossRef]
- Kordesch, K.; Simader, G. Fuel Cells and Their Applications; VCH: Weinheim, New York, NY, USA, 1996; p. 363. [Google Scholar]
- Hydrogen from ammonia. Available online: http://www.acta-nanotech.com (accessed on February 2009).
- Cimenti, M.; Hill, J.M. Direct Utilization of Ethanol on Ceria-Based Anodes for Solid Oxide Fuel Cells. Asian J. Chem. 2009, 4, 45–54. [Google Scholar] [CrossRef]
- Cairns, E.J.; Tevebaugh, A.D.; Holm, G.J. Thermodynamics of Hydrocarbon Fuel Cells. J. Electrochem. Soc. 1963, 110, 1025–1030. [Google Scholar] [CrossRef]
- Sasaki, K.; Teraoka, Y. Equilibria in Fuel Cell Gases - I. Equilibrium Compositions and Reforming Conditions. J. Electrochem. Soc. 2003, 150, A878–A884. [Google Scholar]
- Sasaki, K.; Teraoka, Y. Equilibria in Fuel Cell Gases - II. The C-H-O Ternary Diagrams. J. Electrochem. Soc. 2003, 150, A885–A888. [Google Scholar] [CrossRef]
- Cimenti, M.; Hill, J.M. Thermodynamic Analysis of Solid Oxide Fuel Cells Operated with Methanol and Ethanol under Direct Utilization, Steam Reforming, Dry Reforming or Partial Oxidation Conditions. J. Power Sources 2009, 186, 377–384. [Google Scholar] [CrossRef]
- Walters, K.M.; Dean, A.M.; Zhu, H.; Kee, R.J. Homogeneous Kinetics and Equilibrium Predictions of Coking Propensity in the Anode Channels of Direct Oxidation Solid-Oxide Fuel Cells Using Dry Natural Gas. J. Power Sources 2003, 123, 182–189. [Google Scholar] [CrossRef]
- Sheng, C.Y.; Dean, A.M. Importance of Gas-Phase Kinetics within the Anode Channel of a Solid-Oxide Fuel Cell. J. Phys. Chem. A 2004, 108, 3772–3783. [Google Scholar] [CrossRef]
- Hecht, E.S.; Gupta, G.K.; Zhu, H.Y.; Dean, A.M.; Kee, R.J.; Maier, L.; Deutschmann, O. Methane Reforming Kinetics within a Ni-YSZ SOFC Anode Support. Appl. Catal. A-Gen. 2005, 295, 40–51. [Google Scholar] [CrossRef]
- Gupta, G.K.; Marda, J.R.; Dean, A.M.; Colclasure, A.M.; Zhu, H.Y.; Kee, R.J. Performance Predictions of a Tubular SOFC Operating on a Partially Reformed JP-8 Surrogate. J. Power Sources 2006, 162, 553–562. [Google Scholar] [CrossRef]
- Randolph, K.L.; Dean, A.M. Hydrocarbon Fuel Effects in Solid-Oxide Fuel Cell Operation: An Experimental and Modeling Study of n-Hexane Pyrolysis. Phys. Chem. Chem. Phys. 2007, 9, 4245–4258. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.K.; Dean, A.M.; Ahn, K.; Gorte, R.J. Comparison of Conversion and Deposit Formation of Ethanol and Butane under SOFC Conditions. J. Power Sources 2006, 158, 497–503. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R.; Sehested, J.; Norskov, J.K. Hydrogen and Synthesis Gas by Steam- and CO2 Reforming. In Advances In Catalysis; Academic Press: London, UK, 2002; Vol 47, pp. 65–139. [Google Scholar]
- Rostrup-Nielsen, J.R.; Rostrup-Nielsen, T. Large-Scale Hydrogen Production. Cattech 2002, 6, 150–159. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R. Conversion of Hydrocarbons and Alcohols for Fuel Cells. Phys. Chem. Chem. Phys. 2001, 3, 283–288. [Google Scholar] [CrossRef]
- Peppley, B.A.; Amphlett, J.C.; Kearns, L.M.; Mann, R.F. Methanol-Steam Reforming on Cu/ZnO/Al2O3. Part 1: The Reaction Network. Appl. Catal. A-Gen. 1999, 179, 21–29. [Google Scholar]
- Peppley, B.A.; Amphlett, J.C.; Kearns, L.M.; Mann, R.F. Methanol-steam Reforming on Cu/ZnO/Al2O3 Catalysts. Part 2. A Comprehensive Kinetic Model. Appl. Catal. A-Gen. 1999, 179, 31–49. [Google Scholar] [CrossRef]
- Amphlett, J.C.; Mann, R.F.; Peppley, B.A. The Steam Reforming of Methanol - Mechanism and Kinetics Compared to the Methanol Synthesis Process. In Natural Gas Conversion II; CurryHyde, H.E., Howe, R.F., Eds.; 1994; Vol 81, pp. 409–411. [Google Scholar]
- Cimenti, M.; Hill, J.M. Importance of Pyrolysis and Catalytic Decomposition for the Direct Utilization of Methanol in Solid Oxide Fuel Cells. J. Power Sources 2009. submitted. [Google Scholar] [CrossRef]
- Turns, S.R. An introduction to combustion - concepts and applications, 2nd ed.; McGraw-Hill International Editions: Boston, MA, USA, 2000; p. 676. [Google Scholar]
- Bartholomew, C.H. Carbon Deposition in Steam Reforming and Methanation. Catal. Rev-Sci. Eng. 1982, 24, 67–112. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Catalyst Deactivation. Chem. Eng. 1984, 91, 96–112. [Google Scholar]
- Bartholomew, C.H. Mechanisms of Catalyst Deactivation. Appl. Catal. A-Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Trimm, D.L. Coke Formation and Minimisation during Steam Reforming Reactions. Catal. Today 1997, 37, 233–238. [Google Scholar] [CrossRef]
- Trimm, D.L. Catalysts for the Control of Coking during Steam Reforming. Catal. Today 1999, 49, 3–10. [Google Scholar] [CrossRef]
- He, H.; Hill, J.M. Carbon Deposition on Ni/YSZ Composites Exposed to Humidified Methane. Appl. Catal. A-Gen. 2007, 317, 284–292. [Google Scholar]
- Nikooyeh, K.; Clemmer, R.; Alzate-Restrepo, V.; Hill, J.M. Effect of Hydrogen on Carbon Formation on Ni/YSZ Composites Exposed to Methane. Appl. Catal. A-Gen. 2008, 347, 106–111. [Google Scholar] [CrossRef]
- Alzate-Restrepo, V.; Hill, J.M. Effect of Anodic Polarization on Carbon Deposition on Ni/YSZ Anodes Exposed to Methane. Appl. Catal. A-Gen. 2008, 342, 49–55. [Google Scholar] [CrossRef]
- Weber, A.; Sauer, B.; Muller, A.C.; Herbstritt, D.; Ivers-Tiffee, E. Oxidation of H2, CO and Methane in SOFCs with Ni/YSZ-Cermet Anodes. Solid State Ionics 2002, 152, 543–550. [Google Scholar] [CrossRef]
- Mcintosh, S.; Vohs, J.M.; Gorte, R.J. Role of Hydrocarbon Deposits in the Enhanced Performance of Direct-Oxidation SOFCs. J. Electrochem. Soc. 2003, 150, A470–A476. [Google Scholar] [CrossRef]
- Reinhold, H.; Hans-Heinrich, M. On a Thermodynamic Limitation of the Water-Gas Potentiometry Using Solid Electrolyte Cells. Fresenius J. Anal. Chem. 1994, 349, 571–574. [Google Scholar]
- Atkinson, A.; Barnett, S.; Gorte, R.J.; Irvine, J.T.S.; Mcevoy, A.J.; Mogensen, M.; Singhal, S.C.; Vohs, J. Advanced Anodes for High-Temperature Fuel Cells. Nat. Mater. 2004, 3, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.D.; Vohs, J.M.; Gorte, R.J. Recent Progress in SOFC Anodes for Direct Utilization of Hydrocarbons. J. Mater. Chem. 2007, 17, 3071–3077. [Google Scholar] [CrossRef]
- Zhu, W.Z.; Deevi, S.C. A Review on the Status of Anode Materials for Solid Oxide Fuel Cells. Mater. Sci. Eng. A 2003, 362, 228–239. [Google Scholar] [CrossRef]
- Gorte, R.J.; Vohs, J.M.; Mcintosh, S. Recent Developments on Anodes for Direct Fuel Utilization in SOFC. Solid State Ionics 2004, 175, 1–6. [Google Scholar] [CrossRef]
- Jiang, S.P.; Chan, S.H. A Review of Anode Materials Development in Solid Oxide Fuel Cells. J. Mater. Sci. 2004, 39, 4405–4439. [Google Scholar] [CrossRef]
- Fergus, J.W. Oxide Anode Materials for Solid Oxide Fuel Cells. Solid State Ionics 2006, 177, 1529–1541. [Google Scholar] [CrossRef]
- Liu, J. Direct-Hydrocarbon Solid Oxide Fuel Cells. Prog. Chem. 2006, 18, 1026–1033. [Google Scholar]
- Goodenough, J.B.; Huang, Y.-H. Alternative Anode Materials for Solid Oxide Fuel Cells. J. Power Sources 2007, 173, 1–10. [Google Scholar] [CrossRef]
- Fergus, J.W.; Hui, R.; Li, X.; Wilkinson, D.P.; Zhang, J. Solid oxide fuel cells: Materials Properties and Performance; CRC Press: Boca Raton, FL, USA, 2009; p. 295. [Google Scholar]
- Sun, C.W.; Stimming, U. Recent Anode Advances in Solid Oxide Fuel Cells. J. Power Sources 2007, 171, 247–260. [Google Scholar] [CrossRef]
- Lin, Y.B.; Zhan, Z.L.; Liu, J.; Barnett, S.A. Direct Operation of Solid Oxide Fuel Cells with Methane Fuel. Solid State Ionics 2005, 176, 1827–1835. [Google Scholar] [CrossRef]
- Proctor, I.A.; Hopkin, A.L.; Ormerod, R.M. Development of Anodes for Direct Electrocatalytic Oxidation of Methane in Solid Oxide Fuel Cells. Ionics 2003, 9, 242–247. [Google Scholar] [CrossRef]
- Ilias, G.; Stylianos, N. High Tolerant to Carbon Deposition Ni-based Electrodes under Internal Steam Reforming Conditions. ECS Transactions 2007, 7, 1483–1490. [Google Scholar]
- Nikolla, E.; Schwank, J.; Linic, S. Promotion of the Long-Term Stability of Reforming Ni Catalysts by Surface Alloying. J. Catal. 2007, 250, 85–93. [Google Scholar] [CrossRef]
- Finnerty, C.M.; Cunningham, R.H.; Ormerod, R.M. Study of the Catalysis and Surface Chemistry Occurring at Nickel/Zirconia Anodes in Solid Oxide Fuel Cells Running on Natural Gas. Radiat. Eff. Defect. Solid. 1999, 151, 77–81. [Google Scholar] [CrossRef]
- Eguchi, K.; Kojo, H.; Takeguchi, T.; Kikuchi, R.; Sasaki, K. Fuel Flexibility in Power Generation by Solid Oxide Fuel Cells. Solid State Ionics 2002, 152, 411–416. [Google Scholar] [CrossRef]
- Sumi, H.; Ukai, K.; Mizutani, Y.; Mori, H.; Wen, C.J.; Takahashi, H.; Yamamoto, O. Performance of Nickel-Scandia-Stabilized Zirconia Cermet Anodes for SOFCs in 3% H2O-CH4. Solid State Ionics 2004, 174, 151–156. [Google Scholar] [CrossRef]
- Gunji, A.; Wen, C.; Otomo, J.; Kobayashi, T.; Ukai, K.; Mizutani, Y.; Takahashi, H. Carbon Deposition Behaviour on Ni-ScSZ Anodes for Internal Reforming Solid Oxide Fuel Cells. J. Power Sources 2004, 131, 285–288. [Google Scholar] [CrossRef]
- Kishimoto, H.; Horita, T.; Yamaji, K.; Xiong, Y.; Sakai, N.; Yokokawa, H. Attempt of Utilizing Liquid Fuels with Ni-ScSZ Anode in SOFCs. Solid State Ionics 2004, 175, 107–111. [Google Scholar] [CrossRef]
- Putna, E.S.; Stubenrauch, J.; Vohs, J.M.; Gorte, R.J. Ceria-Based Anodes for the Direct Oxidation of Methane in Solid Oxide Fuel Cells. Langmuir 1995, 11, 4832–4837. [Google Scholar] [CrossRef]
- Murray, E.P.; Tsai, T.; Barnett, S.A. A Direct-Methane Fuel Cell with a Ceria-Based Anode. Nature 1999, 400, 649–651. [Google Scholar] [CrossRef] [PubMed]
- Perry Murray, E. Solid Oxide Fuel Cells Designed for Low - Temperature Operation and Hydrocarbon Fuel Utilization. Ph.D. Thesis, Northwestern University, Evanston, IL, USA, 1999.
- Ormerod, R.M. Solid Oxide Fuel Cells. Chem. Soc. Rev. 2003, 32, 17–28. [Google Scholar] [CrossRef]
- Park, S.; Craciun, R.; Vohs, J.M.; Gorte, R.J. Direct Oxidation of Hydrocarbons in a Solid Oxide Fuel Cell I. Methane Oxidation. J. Electrochem. Soc. 1999, 146, 3603–3605. [Google Scholar] [CrossRef]
- Craciun, R.; Park, S.; Gorte, R.J.; Vohs, J.M.; Wang, C.; Worrell, W.L. Novel Method for Preparing Anode Cermets for Solid Oxide Fuel Cells. J. Electrochem. Soc. 1999, 146, 4019–4022. [Google Scholar] [CrossRef]
- Park, S.; Gorte, R.J.; Vohs, J.M. Tape Cast Solid Oxide Fuel Cells for the Direct Oxidation of Hydrocarbons. J. Electrochem. Soc. 2001, 148, A443–A447. [Google Scholar] [CrossRef]
- Kim, H.; Vohs, J.M.; Gorte, R.J. Direct Oxidation of Sulfur-Containing Fuels in a Solid Oxide Fuel Cell. Chem. Commun. 2001, 22, 2334–2335. [Google Scholar] [CrossRef]
- Park, S.D.; Vohs, J.M.; Gorte, R.J. Direct Oxidation of Hydrocarbons in a Solid-Oxide Fuel Cell. Nature 2000, 404, 265–267. [Google Scholar] [PubMed]
- Park, S.; Gorte, R.J.; Vohs, J.M. Applications of Heterogeneous Catalysis in the Direct Oxidation of Hydrocarbons in a Solid-Oxide Fuel Cell. Appl. Catal. A-Gen. 2000, 200, 55–61. [Google Scholar] [CrossRef]
- Mcintosh, S.; Vohs, J.M.; Gorte, R.J. An Examination of Lanthanide Additives on the Performance of Cu-YSZ Cermet anodes. Electrochim. Acta 2002, 47, 3815–3821. [Google Scholar] [CrossRef]
- Lu, C.; Worrell, W.L.; Vohs, J.M.; Gorte, R.J. A Comparison of Cu-ceria-SDC and Au-ceria-SDC Composites for SOFC Anodes. J. Electrochem. Soc. 2003, 150, A1357–A1359. [Google Scholar] [CrossRef]
- Marina, O.A.; Bagger, C.; Primdahl, S.; Mogensen, M. A Solid Oxide Fuel Cell with a Gadolinia-Doped Ceria Anode: Preparation and Performance. Solid State Ionics 1999, 123, 199–208. [Google Scholar] [CrossRef]
- Marina, O.A.; Mogensen, M. High-Temperature Conversion of Methane on a Composite Gadolinia-Doped Ceria-Gold Electrode. Appl. Catal. A-Gen. 1999, 189, 117–126. [Google Scholar] [CrossRef]
- Lu, C.; Worrell, W.L.; Gorte, R.J.; Vohs, J.M. SOFCs for Direct Oxidation of Hydrocarbon Fuels with Samaria-Doped Ceria Electrolyte. J. Electrochem. Soc. 2003, 150, A354–A358. [Google Scholar] [CrossRef]
- Costa-Nunes, O.; Gorte, R.J.; Vohs, J.M. High Mobility of Ceria Films on Zirconia at Moderate Temperatures. J. Mater. Chem. 2005, 15, 1520–1522. [Google Scholar] [CrossRef]
- Kim, H.; Lu, C.; Worrell, W.L.; Vohs, J.M.; Gorte, R.J. Cu-Ni Cermet Anodes for Direct Oxidation of Methane in Solid-Oxide Fuel Cells. J. Electrochem. Soc. 2002, 149, A247–A250. [Google Scholar] [CrossRef]
- Lee, S.I.; Vohs, J.M.; Gorte, R.J. A Study of SOFC Anodes Based on Cu-Ni and Cu-Co Bimetallics in CeO2-YSZ. J. Electrochem. Soc. 2004, 151, A1319–A1323. [Google Scholar] [CrossRef]
- Lee, S.I.; Ahn, K.; Vohs, J.M.; Gorte, R.J. Cu-Co Bimetallic Anodes for Direct Utilization of Methane in SOFCs. Electrochem. Solid State Lett. 2005, 8, A48–A51. [Google Scholar] [CrossRef]
- Mcintosh, S.; Vohs, J.M.; Gorte, R.J. Effect of Precious-Metal Dopants on SOFC Anodes for Direct Utilization of Hydrocarbons. Electrochem. Solid State Lett. 2003, 6, A240–A243. [Google Scholar] [CrossRef]
- Jung, S.; Gross, M.D.; Gorte, R.J.; Vohs, J.M. Electrodeposition of Cu into a Highly Porous Ni/YSZ Cermet. J. Electrochem. Soc. 2006, 153, A1539–A1543. [Google Scholar] [CrossRef]
- Gross, M.D.; Vohs, J.M.; Gorte, R.J. Enhanced Thermal Stability of Cu-Based SOFC Anodes by Electrodeposition of Cr. J. Electrochem. Soc. 2006, 153, A1386–A1390. [Google Scholar] [CrossRef]
- Gross, M.D.; Vohs, J.M.; Gorte, R.J. A study of Thermal Stability and Methane Tolerance of Cu-Based SOFC Anodes with Electrodeposited Co. Electrochim. Acta 2007, 52, 1951–1957. [Google Scholar] [CrossRef]
- Gross, M.D.; Vohs, J.M.; Gorte, R.J. A Strategy for Achieving High Performance with SOFC Ceramic Anodes. Electrochem. Solid State Lett. 2007, 10, B65–B69. [Google Scholar] [CrossRef]
- Gross, M.D.; Vohs, J.M.; Gorte, R.J. An Examination of SOFC Anode Functional Layers Based on Ceria in YSZ. J. Electrochem. Soc. 2007, 154, B694–B699. [Google Scholar] [CrossRef]
- Pillai, M.R.; Kim, I.; Bierschenk, D.M.; Barnett, S.A. Fuel-Flexible Operation of a Solid Oxide Fuel Cell with Sr0.8La0.2TiO3 Support. J. Power Sources 2008, 185, 1086–1093. [Google Scholar] [CrossRef]
- Tao, S.W.; Irvine, J.T.S. A Redox-Stable Efficient Anode for Solid-Oxide Fuel Cells. Nat. Mater. 2003, 2, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Zha, S.W.; Tsang, P.; Cheng, Z.; Liu, M.L. Electrical Properties and Sulfur Tolerance of La0.75Sr0.25Cr1-xMnxO3 under Anodic Conditions. J. Solid State Chem. 2005, 178, 1844–1850. [Google Scholar] [CrossRef]
- Tao, S.W.; Irvine, J.T.S. Phase Transition in Perovskite Oxide La0.75Sr0.25Cr0.5Mn0.5O3-δ Observed by in situ High-Temperature Neutron Powder Diffraction. Chem. Mater. 2006, 18, 5453–5460. [Google Scholar]
- Tao, S.W.; Irvine, J.T.S.; Plint, S.M. Methane Oxidation at Redox Stable Fuel Cell Electrode La0.75Sr0.25Cr0.5Mn0.5O3-δ. J. Phys. Chem. B 2006, 110, 21771–21776. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Morales, J.C.; Canales-Vazquez, J.; Ballesteros-Perez, B.; Pena-Martinez, J.; Marrero-Lopez, D.; Irvine, J.T.S.; Nunaz, P. LSCM-(YSZ-CGO) Composites as Improved Symmetrical Electrodes for Solid Oxide Fuel Cells. J. Eur. Ceram. Soc. 2007, 27, 4223–4227. [Google Scholar] [CrossRef]
- Ruiz-Morales, J.C.; Canales-Vazquez, J.; Pena-Martinez, J.; Lopez, D.M.; Nunez, P. On the Simultaneous Use of La0.75Sr0.25Cr0.5Mn0.5O3-δ as Both Anode and Cathode Material with Improved Microstructure in Solid Oxide Fuel Cells. Electrochim. Acta 2006, 52, 278–284. [Google Scholar]
- Pena-Martinez, J.; Marrero-Lopez, D.; Ruiz-Morales, J.C.; Buergler, B.E.; Nunez, P.; Gauckler, L.J. Fuel Cell Studies of Perovskite-Type Materials for IT-SOFC. J. Power Sources 2006, 159, 914–921. [Google Scholar] [CrossRef]
- Bastidas, D.M.; Tao, S.W.; Irvine, J.T.S. A Symmetrical Solid Oxide Fuel Cell Demonstrating Redox Stable Perovskite Electrodes. J. Mater. Chem. 2006, 16, 1603–1605. [Google Scholar] [CrossRef]
- Cheng, Z.; Zha, S.; Aguilar, L.; Wang, D.; Winnick, J.; Liu, M. A Solid Oxide Fuel Cell Running on H2S/CH4 Fuel Mixtures. Electrochem. Solid-State Lett. 2006, 9, A31–A33. [Google Scholar] [CrossRef]
- Jiang, S.P.; Chen, X.J.; Chan, S.H.; Kwok, J.T. GDC-Impregnated, (La0.75Sr0.25)(Cr0.5Mn0.5)O3 Anodes for Direct Utilization of Methane in Solid Oxide Fuel Cells. J. Electrochem. Soc. 2006, 153, A850–A856. [Google Scholar]
- Ruiz-Morales, J.C.; Canales-Vazquez, J.; Marrero-Lopez, D.; Irvine, J.T.S.; Nunez, P. Improvement of the Electrochemical Properties of Novel Solid Oxide Fuel Cell Anodes, La0.75Sr0.25Cr0.5Mn0.5O3-d and La4Sr8Ti11Mn0.5Ga0.5O37.5-δ, Using Cu-YSZ-Based Cermets. Electrochim. Acta 2007, 52, 7217–7225. [Google Scholar]
- Hui, S.Q.; Petric, A. Evaluation of Yttrium-doped SrTiO3 as an Anode for Solid Oxide Fuel Cells. J. Eur. Ceram. Soc. 2002, 22, 1673–1681. [Google Scholar] [CrossRef]
- Hui, S. Evaluation of Yttrium-doped SrTiO3 as a Solid Oxide Fuel Cell Anode. Ph.D. Thesis, McMaster University, Hamilton, Canada, 2000. [Google Scholar]
- He, H.; Huang, Y.; Vohs, J.M.; Gorte, R.J. Characterization of YSZ-YST Composites for SOFC Anodes. Solid State Ionics 2004, 175, 171–176. [Google Scholar] [CrossRef]
- Mukundan, R.; Brosha, E.L.; Garzon, F.H. Sulfur Tolerant Anodes for SOFCs. Electrochem. Solid-State Lett. 2004, 7, A5–A7. [Google Scholar] [CrossRef]
- Aguilar, L.; Zha, S.; Cheng, Z.; Winnick, J.; Liu, M. A Solid Oxide Fuel Cell Operating on Hydrogen Sulfide (H2S) and Sulfur-Containing Fuels. J. Power Sources 2004, 135, 17–24. [Google Scholar] [CrossRef]
- Aguilar, L.; Zha, S.; Li, S.; Winnick, J.; Liu, M. Sulfur-Tolerant Materials for the Hydrogen Sulfide SOFC. Electrochem. Solid-State Lett. 2004, 7, A324–A326. [Google Scholar] [CrossRef]
- Huang, Y.H.; Dass, R.I.; Denyszyn, J.C.; Goodenough, J.B. Synthesis and Characterization of Sr2MgMoO6-δ - An Anode Material for the Solid Oxide Fuel Cell. J. Electrochem. Soc. 2006, 153, A1266–A1272. [Google Scholar] [CrossRef]
- Huang, Y.H.; Dass, R.I.; Xing, Z.L.; Goodenough, J.B. Double Perovskites as Anode Materials for Solid-Oxide Fuel Cells. Science 2006, 312, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Huang, Y.H.; Ying, J.R.; Goodenough, J.B. Electrochemical Performance of La-doped Sr2MgMoO6-δ in Natural Gas. Electrochem. Commun. 2007, 9, 1881–1885. [Google Scholar] [CrossRef]
- Sammes, N.; Varadaraj, L. Methanol Oxidation Over Doped-LaCoO3 Electrodes in a Solid Oxide Fuel-Cell. Denki Kagaku 1995, 63, 41–46. [Google Scholar]
- Jiang, Y.; Virkar, A.V. A High Performance, Anode-Supported Solid Oxide Fuel Cell Operating on Direct Alcohol. J. Electrochem. Soc. 2001, 148, A706–A709. [Google Scholar] [CrossRef]
- Brett, D.J.L.; Atkinson, A.; Cumming, D.; Ramirez-Cabrera, E.; Rudkin, R.; Brandon, N.P. Methanol as a Direct Fuel in Intermediate Temperature (500-) Solid Oxide Fuel Cells with Copper Based Anodes. Chem. Eng. Science 2005, 60, 5649–5662. [Google Scholar] [CrossRef]
- Kim, T.; Ahn, K.; Vohs, J.M.; Gorte, R.J. Deactivation of Ceria-Based SOFC Anodes in Methanol. J. Power Sources 2007, 164, 42–48. [Google Scholar] [CrossRef]
- Zhu, B.; Liu, X.; Zhou, P.; Yang, X.; Zhu, Z.; Zhu, W. Innovative Solid Carbonate-Ceria Composite Electrolyte Fuel Cells. Electrochem. Commun. 2001, 3, 566–571. [Google Scholar] [CrossRef]
- Xu, S.H.; Niu, X.M.; Chen, M.M.; Wang, C.Y.; Zhu, B. Carbon Doped MO-SDC Material as an SOFC Anode. J. Power Sources 2007, 165, 82–86. [Google Scholar] [CrossRef]
- Mat, M.D.; Liu, X.R.; Zhu, Z.G.; Zhu, B. Development of Cathodes for Methanol and Ethanol Fuelled Low Temperature (300-600 degrees C) Solid Oxide Fuel Cells. Int. J. Hydrogen. Energ. 2007, 32, 796–801. [Google Scholar] [CrossRef]
- Sasaki, K.; Watanabe, K.; Teraoka, Y. Direct-Alcohol SOFCs: Current-Voltage Characteristics and Fuel Gas Compositions. J. Electrochem. Soc. 2004, 151, A965–A970. [Google Scholar] [CrossRef]
- Huang, B.; Wang, S.R.; Liu, R.Z.; Ye, X.E.; Nie, H.W.; Sun, X.E.; Wen, T.L. Performance of La0.75Sr0.25Cr0.5Mn0.5O3-δ Perovskite-Structure Anode Material at Lanthanum Gallate Electrolyte for IT-SOFC Running on Ethanol Fuel. J. Power Sources 2007, 167, 39–46. [Google Scholar]
- Ye, X.F.; Huang, B.; Wang, S.R.; Wang, Z.R.; Xiong, L.; Wen, T.L. Preparation and Performance of a Cu-CeO2-ScSZ Composite Anode for SOFCs Running on Ethanol Fuel. J. Power Sources 2007, 164, 203–209. [Google Scholar] [CrossRef]
- Huang, B.; Wang, S.R.; Liu, R.Z.; Wen, T.L. Preparation and Performance Characterization of the Fe-Ni/ScSZ Cermet Anode for Oxidation of Ethanol Fuel in SOFCs. J. Power Sources 2007, 167, 288–294. [Google Scholar] [CrossRef]
- Cimenti, M. Direct Utilization of Methanol and Ethanol in Solid Oxide Fuel Cells. Ph.D. Thesis, University of Calgary, Calgary, Canada, 2008. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cimenti, M.; Hill, J.M. Direct Utilization of Liquid Fuels in SOFC for Portable Applications: Challenges for the Selection of Alternative Anodes. Energies 2009, 2, 377-410. https://doi.org/10.3390/en20200377
Cimenti M, Hill JM. Direct Utilization of Liquid Fuels in SOFC for Portable Applications: Challenges for the Selection of Alternative Anodes. Energies. 2009; 2(2):377-410. https://doi.org/10.3390/en20200377
Chicago/Turabian StyleCimenti, Massimiliano, and Josephine M. Hill. 2009. "Direct Utilization of Liquid Fuels in SOFC for Portable Applications: Challenges for the Selection of Alternative Anodes" Energies 2, no. 2: 377-410. https://doi.org/10.3390/en20200377