
A Data-Based Hybrid Chemistry Acceleration Framework for the Low-Temperature Oxidation of Complex Fuels

Abstract
:1. Introduction
2. Methodology
2.1. Database Generation
- The energy equation
- The species equation
- The enthalpy equation
- The species equation
2.2. Representative Species Selection
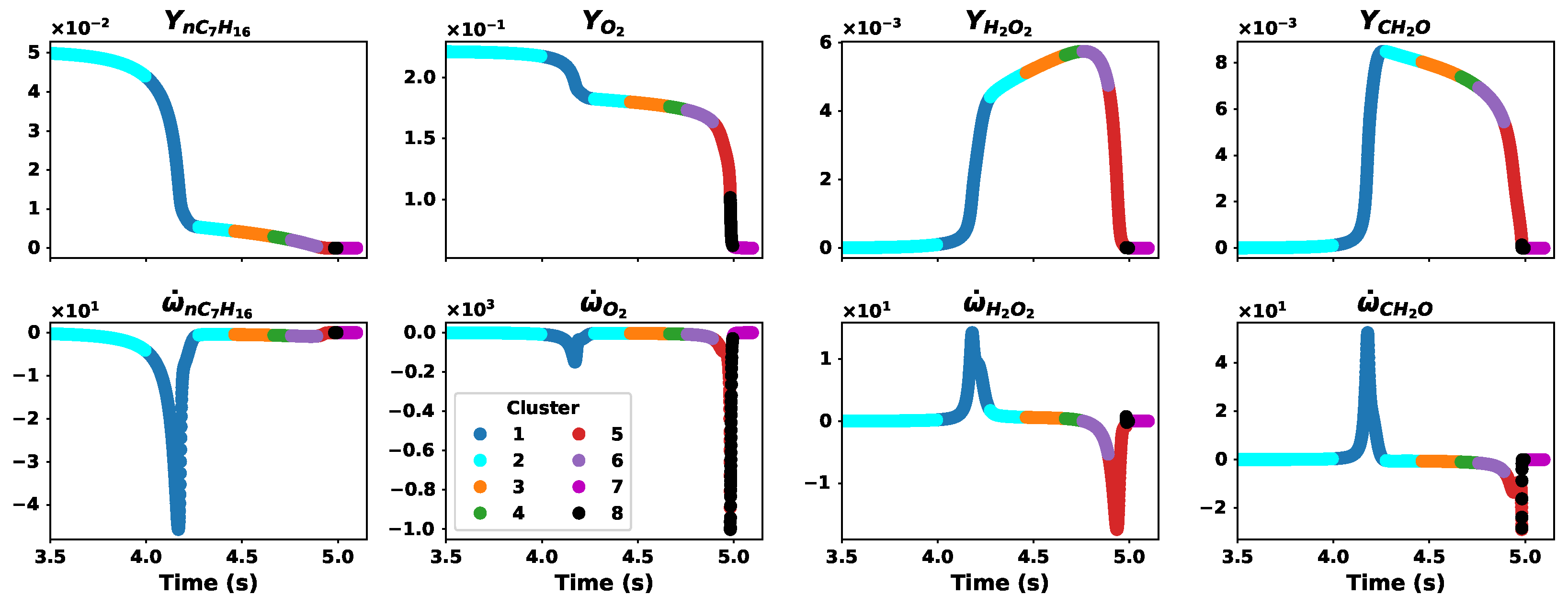
2.3. Data Clustering
- (a)
- Using the offset percentage from zero , we obtain the offsetwhere is the reaction rate PCs;
- (b)
- The cluster borders are obtained using the offset aswhere and are the negative and positive borders of a cluster, is the set number of clusters, is the absolute value operator, and is the function to evenly space numbers over a specified interval ();
- (c)
- Data are then clustered based on where they fall along the negative or positive borders.
2.4. Artificial Neural Network Architecture
- Input scalingwhere
- Reaction rate scaling
2.5. Summary of the Hybrid Chemistry Model
- The energy equation
- The representative species equation
- Remaining species equation
3. Results and Discussion
3.1. Selected Representative Species
3.2. Data Clusters
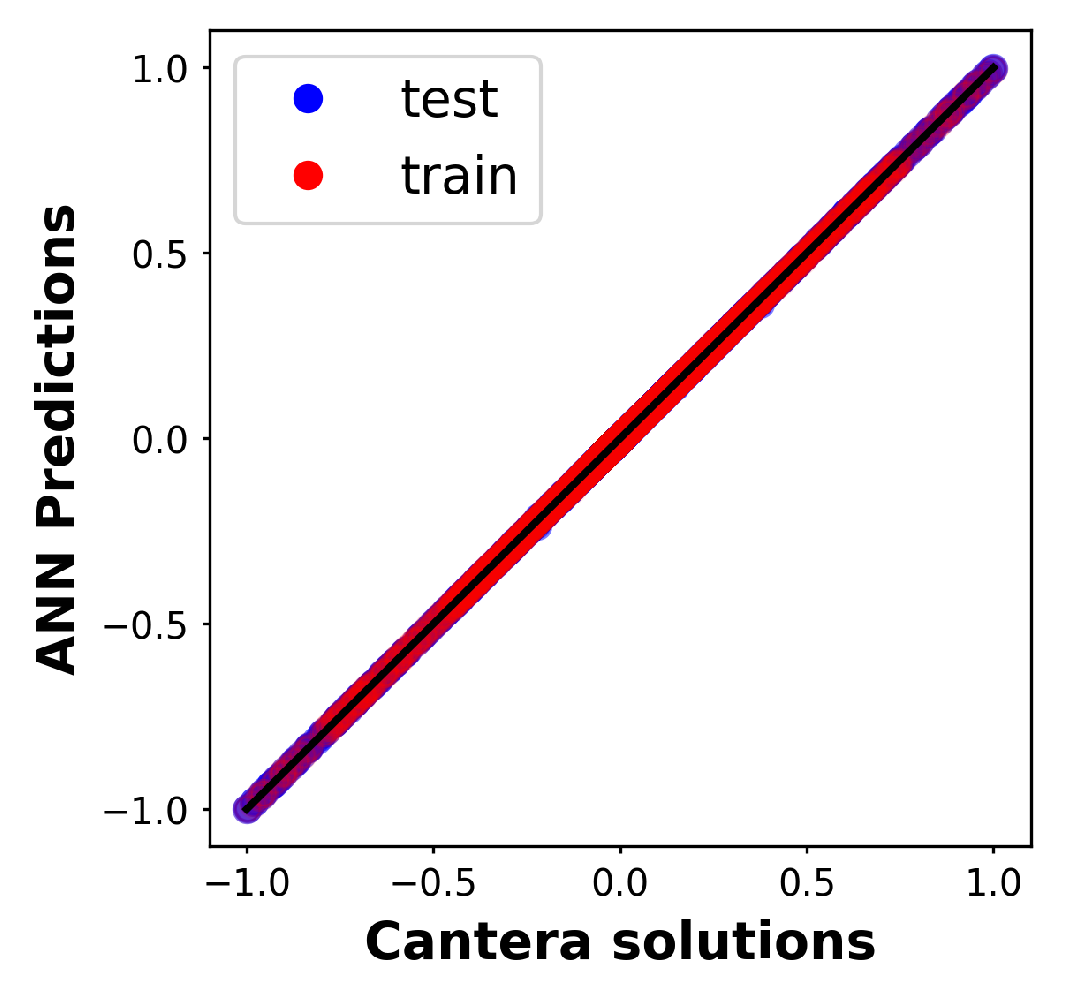
3.3. ANN Training and Reaction Rate Prediction
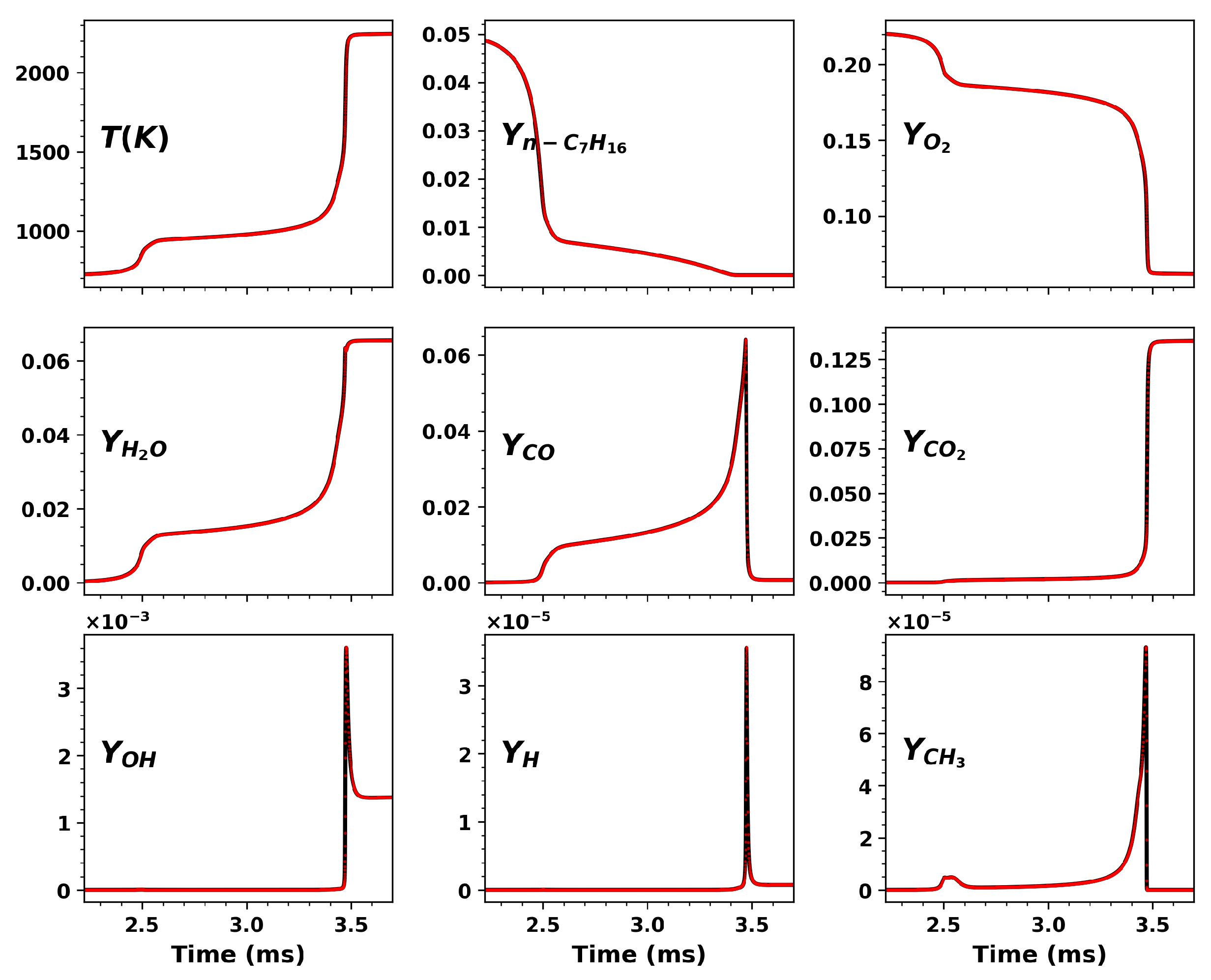
3.4. Hybrid Chemistry Model
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, H.; Xu, R.; Wang, K.; Bowman, C.; Hanson, R.; Davidson, D.; Brezinsky, K. A physics-based approach to modeling real-fuel combustion chemistry-I. evidence from experiments, and thermodynamic, chemical kinetic and statistical considerations. Combust. Flame 2018, 193, 502–519. [Google Scholar] [CrossRef]
- Xu, R.; Wang, K.; Banerjee, S.; Shao, J.; Parise, T.; Zhu, Y.; Wang, S.; Movaghar, A.; Lee, D.; Zhao, R.; et al. A physics-based approach to modeling real-fuel combustion chemistry-II. reaction kinetic models of jet and rocket fuels. Combust. Flame 2018, 193, 520–537. [Google Scholar] [CrossRef]
- Tao, Y.; Xu, R.; Wang, K.; Shao, J.; Johnson, S.; Movaghar, A.; Han, X.; Park, J.-W.; Lu, T.; Brezinsky, K.; et al. A physics-based approach to modeling real-fuel combustion chemistry-III. reaction kinetic model for JP10. Combust. Flame 2018, 198, 466–476. [Google Scholar] [CrossRef]
- Zádor, J.; Taatjes, C.; Fernandes, R. Kinetics of elementary reactions in low-temperature autoignition chemistry. Prog. Energy Combust. Sci. 2011, 37, 371–421. [Google Scholar] [CrossRef]
- Alqahtani, S.; Echekki, T. A data-based hybrid model for complex fuel chemistry acceleration at high temperatures. Combust. Flame 2021, 223, 142–152. [Google Scholar] [CrossRef]
- Ranade, R.; Alqahtani, S.; Farooq, A.; Echekki, T. An ANN based hybrid chemistry framework for complex Fuels. Fuel 2019, 241, 625–636. [Google Scholar] [CrossRef]
- Ranade, R.; Echekki, T. An extended hybrid chemistry framework for complex hydrocarbon fuels. Fuel 2019, 251, 276–284. [Google Scholar] [CrossRef]
- Brown, N.; Li, G.; Koszykowski, M. Mechanism reduction via principal component analysis. Int. J. Chem. Kin. 1996, 29, 393–414. [Google Scholar] [CrossRef]
- Alqahtani, S. Machine Learning Methods for Chemistry Reduction in Combustion. Ph.D. Thesis, North Carolina State University, Department of Mechanical and Aerospace Engineering, Raleigh, NC, USA, 2020. [Google Scholar]
- Xu, R.; Wang, H.; Davidson, D.F.; Hanson, R.K.; Bowman, C.T.; Egolfopoulos, F.N. Evidence supporting a simplified approach to modeling high-temperature combustion chemistry. In Proceedings of the 10th US National Meeting on Combustion, College Park, MD, USA, 23–24 April 2017. [Google Scholar]
- Xu, R.; Chen, D.; Wang, K.; Tao, Y.; Shao, J.K.; Parise, T.; Zhu, Y.; Wang, S.; Zhao, R.; Lee, D.J.; et al. HyChem model: Application to petroleum-derived jet fuels. In Proceedings of the 10th US National Meeting on Combustion, College Park, MD, USA, 23–24 April 2017; Volume 69, pp. 70–77. [Google Scholar]
- Xu, R.; Chen, D.; Wang, K.; Wang, H. A comparative study of combustion chemistry of conventional and alternative jet fuels with hybrid chemistry approach. In Proceedings of the 55th AIAA Aerospace Sciences Meeting, Grapevine, TX, USA, 9–13 January 2017; p. 0607. [Google Scholar]
- Wang, K.; Xu, R.; Parise, T.; Shao, J.K.; Lee, D.J.; Movaghar, A.; Davidson, D.F.; Hanson, R.K.; Wang, H.; Bowman, C.T.; et al. Combustion kinetics of conventional and alternative jet fuels using a hybrid chemistry (HyChem) approach. In Proceedings of the 10th US National Combustion Institute Meeting, the Combustion Institute, College Park, MD, USA, 23–24 April 2017. [Google Scholar]
- Wang, K.; Xu, R.; Parise, T.; Shao, J.K.; Davidson, D.F.; Hanson, R.K.; Wang, H.; Bowman, C.T. Evaluation of a hybrid chemistry approach for combustion blended petroleum and bio-derived jet fuels. In Proceedings of the 10th US National Combustion Institute Meeting, the Combustion Institute, College Park, MD, USA, 23–24 April 2017. [Google Scholar]
- You, X.; Egolfopoulos, F.N.; Wang, H. Detailed and simplified kinetic models for n-dodecane oxidation: The role of fuel cracking in aliphatic hydrocarbon combustion. Proc. Combust. Inst. 2009, 32, 403–410. [Google Scholar] [CrossRef]
- Choudhary, R.; Boddapati, V.; Clees, S.; Biswas, P.; Shao, J.; Davidson, D.F.; Hanson, R.K. Towards HyChem modeling of kinetics of distillate fuels in the NTC regime. In Proceedings of the Spring Technical Meeting of the Western States Section of the Combustion Institute, Stanford, CA, USA, 21–22 March 2022. [Google Scholar]
- An, J.; He, G.; Luo, K.; Qin, F.; Liu, B. Artificial neural network based chemical mechanisms for computationally efficient modeling of hydrogen/carbon monoxide/kerosene combustion. Int. J. Hydrogen Energy 2020, 45, 29594–29605. [Google Scholar] [CrossRef]
- Haghshenas, M.; Mitra, P.; Santo, N.D.; Schmidt, D.P. Acceleration of chemical kinetics computation with the learned intelligent tabulation (LIT) method. Energies 2021, 14, 7851. [Google Scholar] [CrossRef]
- Nguyen, H.-T.; Domingo, P.; Vervisch, L.; Nguyen, P.-D. Machine learning for integrating combustion chemistry in numerical simulations. Energy AI 2021, 5, 100082. [Google Scholar] [CrossRef]
- Zhou, L.; Song, Y.; Ji, W.; Wei, H. Machine learning for combustion. Energy AI 2022, 7, 100128. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Blasco, J.A.; Fueyo, N.; Dopazo, C. An economical strategy for storage of chemical kinetics: Fitting in situ adaptive tabulation with artificial neural networks. Proc. Combust. Inst. 2000, 28, 115–121. [Google Scholar] [CrossRef]
- Blasco, J.A.; Fueyo, N.; Dopazo, C.; Ballester, J. Modelling the temporal evolution of a reduced combustion chemical system with an artificial neural network. Combust. Flame 1998, 113, 38–52. [Google Scholar] [CrossRef]
- Blasco, J.A.; Fueyo, N.; Larroya, J.C.; Dopazo, C.; Chen, Y.J. A single-step time-integrator of a methane–air chemical system using artificial neural networks. Comput. Chem. Eng. 1999, 23, 1127–1133. [Google Scholar] [CrossRef]
- Ihme, M.; Marsden, A.L.; Pitsch, H. Generation of optimal artificial neural networks using a pattern search algorithm: Application to approximation of chemical systems. Neural Comput. 2008, 20, 573–601. [Google Scholar] [CrossRef]
- Chatzopoulos, A.K.; Rigopoulos, S. A chemistry tabulation approach via rate-controlled constrained equilibrium (RCCE) and artificial neural networks (ANNs), with application to turbulent non-premixed CH4/H2/N2 flames. Proc. Combust. Inst. 2013, 34, 1465–1473. [Google Scholar] [CrossRef]
- Franke, L.L.; Chatzopoulos, A.K.; Rigopoulos, S. Tabulation of combustion chemistry via Artificial Neural Networks (ANNs): Methodology and application to LES-PDF simulation of Sydney flame L. Combust. Flame 2017, 185, 245–260. [Google Scholar] [CrossRef]
- Sinaei, P.; Tabejamaat, S. Large eddy simulation of methane diffusion jet flame with representation of chemical kinetics using artificial neural network. Proc. Inst. Mech. Eng. Part E J. Process Mech. Eng. 2017, 231, 147–163. [Google Scholar] [CrossRef]
- Lutz, A.E.; Kee, R.J.; Miller, J.A. SENKIN: A FORTRAN Program for Predicting Homogeneous Gas Phase Chemical Kinetics with Sensitivity Analysis; Sandia National Laboratories: Livermore, CA, USA, 1988. [Google Scholar]
- Mehl, M.; Pitz, W.J.; Westbrook, C.K.; Curran, H.J. Kinetic modeling of gasoline surrogate components and mixtures under engine conditions. Proc. Combust. Inst. 2011, 33, 193–200. [Google Scholar] [CrossRef]
- Brunialti, S.; Zhang, X.; Faravelli, T.; Frassoldati, A.; Sarathy, S.M. Automatically generated detailed and lumped reaction mechanisms for low- and high-temperature oxidation of alkanes. Proc. Combust. Inst. 2023, 39, 335–344. [Google Scholar] [CrossRef]
- Barwey, S.; Prakash, S.; Hassanaly, M.; Raman, V. Data-driven classification and modeling of combustion regimes in detonation waves. Flow Turbul. Combust. 2021, 106, 1065–1089. [Google Scholar] [CrossRef]
- Ranade, R.; Li, G.; Li, S.; Echekki, T. An efficient machine-learning approach for pdf tabulation in turbulent combustion closure. Combust. Sci. Tech. 2021, 193, 1258–1277. [Google Scholar] [CrossRef]
- Kohonen, T.; Hynninen, J.; Kangas, J.; Laaksonen, J. SOM_PAK: The self-organizing map program package. Tech. Rep. Citeseer 1996, 31, 1996. [Google Scholar]
- MacQueen, J. Some methods for classification and analysis of multivariate observations. In Fifth Berkeley Symposium on Mathematical Statistics and Probability; University of California Press: Berkeley, CA, USA, 1967; Volume 1, pp. 281–297. [Google Scholar]
- D’Alessio, G.; Parente, A.; Stagni, A.; Cuoci, A. Adaptive chemistry via pre-partitioning of composition space and mechanism reduction. Combust. Flame 2020, 211, 68–82. [Google Scholar] [CrossRef]
- Zdybal, K.; Armstrong, E.; Parente, A.; Sutherland, J.C. PCAfold 2.0-Novel tools and algorithms for low-dimensional manifold assessment and optimization. SoftwareX 2023, 23, 101447. [Google Scholar] [CrossRef]
- MathWorks Inc. Matlab Version: 9.13.0 (r2022b). 2022. Available online: https://www.mathworks.com (accessed on 1 December 2022).
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. PyTorch: An Imperative Style, High-Performance Deep Learning Library. In Advances in Neural Information Processing Systems; Curran Associates, Inc.: Kottayam, India, 2019; Volume 32, pp. 8024–8035. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Buras, Z.; Hansen, N.; Taatjes, C.A.; Sheps, L. Prospects and Limitations of Predicting Fuel Ignition Properties from Low-Temperature Speciation Data. Energy Fuels 2022, 36, 3229–3238. [Google Scholar] [CrossRef]
- Buras, Z.J.; Safta, C.; Zádor, J.; Sheps, L. Simulated production of OH, HO2, CH2O, and CO2 during dilute fuel oxidation can predict 1st-stage ignition delays. Combust. Flame 2020, 216, 472–484. [Google Scholar] [CrossRef]
- Wang, H.; You, X.; Joshi, A.; Davis, S.; Laskin, A.; Egolfopoulos, F.; Law, C.K. USC Mech Version II. High-Temperature Combustion Reaction Model of H2/CO/C1-C4 Compounds, Tech. Rep. (May, 2007). Available online: https://ignis.usc.edu:80/Mechanisms/USC-Mech%20II/USC_Mech%20II.htm (accessed on 15 May 2021).
- Goodwin, D.G.; Moffat, H.K.; Schoegl, I.; Speth, R.L.; Webber, B.W. Cantera: An Object-Oriented Software Toolkit for Chemical Kinetics, Thermodynamics, and Transport Processes. Version 2.6.0. 2022. Available online: https://www.cantera.org (accessed on 20 November 2023).
- Lu, T.F.; Law, C.K. A directed relation graph method for mechanism reduction. Proc. Combust. Inst. 2005, 30, 1333–1341. [Google Scholar] [CrossRef]
- Lu, T.F.; Law, C.K. Linear time reduction of large kinetic mechanisms with directed relation graph: N-heptane and iso-octane. Combust. Flame 2006, 144, 24–36. [Google Scholar] [CrossRef]
- Lu, T.F.; Law, C.K. On the applicability of directed relation graphs to the reduction of reaction mechanisms. Combust. Flame 2006, 146, 472–483. [Google Scholar] [CrossRef]
- Gao, X.; Yang, S.; Sun, W. A global pathway selection algorithm for the reduction of detailed chemical kinetic mechanisms. Combust. Flame 2016, 167, 238–247. [Google Scholar] [CrossRef]
- Dikeman, H.E.; Zhang, H.; Yang, S. Stiffness-reduced neural ODE models for data-driven reduced-order modeling of combustion chemical kinetics. In Proceedings of the AIAA SCITECH 2022 Forum, San Diego, CA, USA, 3–7 January 2022; p. 0226. [Google Scholar]
- Owoyele, O.; Pal, P. Chemnode: A neural ordinary differential equations framework for efficient chemical kinetic solvers. Energy AI 2022, 7, 100118. [Google Scholar] [CrossRef]
- Lee, K.; Parish, E.J. Parameterized neural ordinary differential equations: Applications to computational physics problems. Proc. R. Soc. A Math. Phys. Eng. Sci. 2021, 477, 2253. [Google Scholar] [CrossRef]
- Ji, W.; Richter, F.; Gollner, M.J.; Deng, S. Autonomous kinetic modeling of biomass pyrolysis using chemical reaction neural networks. Combust. Flame 2022, 240, 111992. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fuel | n-C7H16 |
|---|---|
| Selected Species (24) | H, O, OH, HO2, H2, H2O, H2O2, O2, CH3, HCO, CH2O, CH3O, CO, CO2, C2H3, C2H4, C2H5, HCCO, CH2CO, CH3CO, CH2CHO, CH3CHO, C3H6, and C2H3 CHO |
| Cluster | Loss |
|---|---|
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alqahtani, S.; Gitushi, K.M.; Echekki, T. A Data-Based Hybrid Chemistry Acceleration Framework for the Low-Temperature Oxidation of Complex Fuels. Energies 2024, 17, 734. https://doi.org/10.3390/en17030734
Alqahtani S, Gitushi KM, Echekki T. A Data-Based Hybrid Chemistry Acceleration Framework for the Low-Temperature Oxidation of Complex Fuels. Energies. 2024; 17(3):734. https://doi.org/10.3390/en17030734
Chicago/Turabian StyleAlqahtani, Sultan, Kevin M. Gitushi, and Tarek Echekki. 2024. "A Data-Based Hybrid Chemistry Acceleration Framework for the Low-Temperature Oxidation of Complex Fuels" Energies 17, no. 3: 734. https://doi.org/10.3390/en17030734