
Pore-Scale Modeling of Methane Hydrate Dissociation Using a Multiphase Micro-Continuum Framework

Abstract
:1. Introduction
2. Mathematical Models
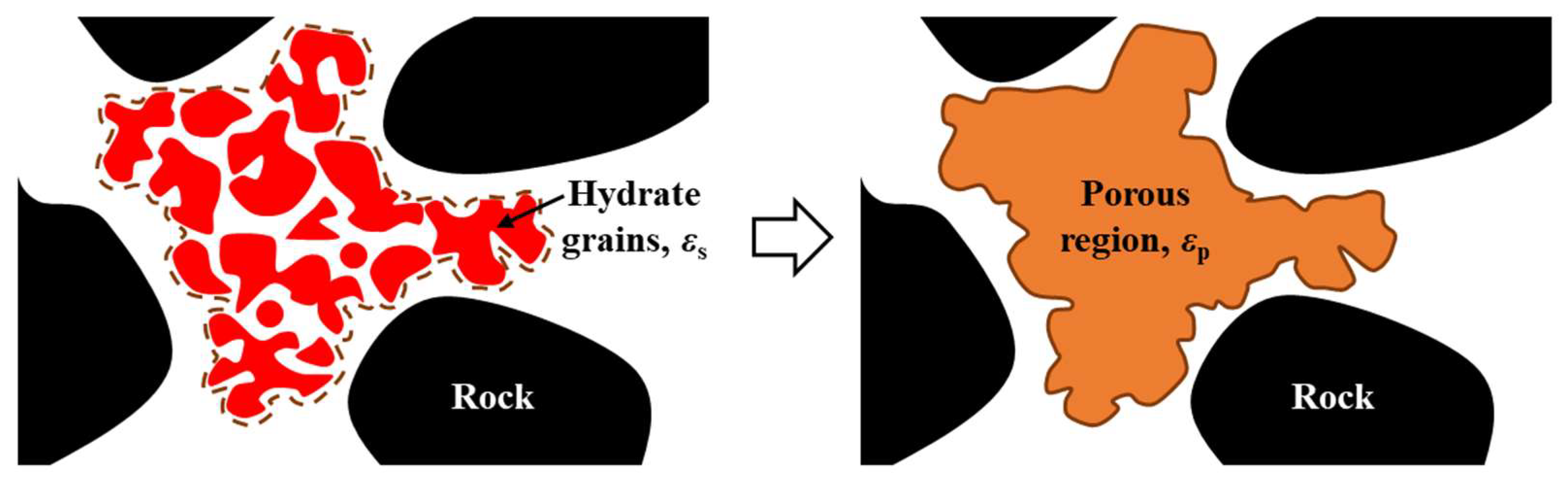
2.1. Hydrate Dissociation Model
2.2. Governing Equations
2.3. Numerical Implementation
- (1)
- The methane hydrate dissolution rate and source terms are calculated based on Equations (A1) and (A2) and with the methane concentration and specific reactive surface area from the previous iteration/time step.
- (2)
- The volume fraction of methane hydrate is explicitly solved by Equation (9). The local porosity and transport properties in the porous matrix are updated.
- (3)
- The saturation equation, Equation (11), is solved using the multidimensional universal limiter with explicit solution (MULES) algorithm to ensure the saturation boundedness.
- (4)
- The normal vector , the curvature of the gas–liquid interface , the surface tension force , the capillary pressure , and single-field transport properties (i.e., ) are updated based on the new saturation field.
- (5)
- The discretized DBS momentum equation, Equation (10), is implicitly solved to obtain the predicted velocity and mass flux , which is the so-called momentum prediction step.
- (6)
- The predicted velocity is corrected based on a predictor–corrector strategy using the splitting of operators (PSIO) algorithms [41], which is the so-called momentum corrector step. The pressure equation is then computed and repeated until the convergence of the pressure and velocity fields. A corrector step number of 2~3 is recommended to guarantee mass conservation.
- (7)
- The concentration equation, Equation (12), is solved using the sequential operator splitting algorithm to improve the numerical stability, wherein the advection term is first solved and then injected as a source term into the diffusion equation [42].
- (8)
- The energy equation, Equation (13), is solved to update the temperature field.
2.4. Numerical Stability
3. Details of Simulation Cases
3.1. Experimental Conditions
3.2. Simulation Configurations
3.3. Mesh Independence
4. Simulation Results
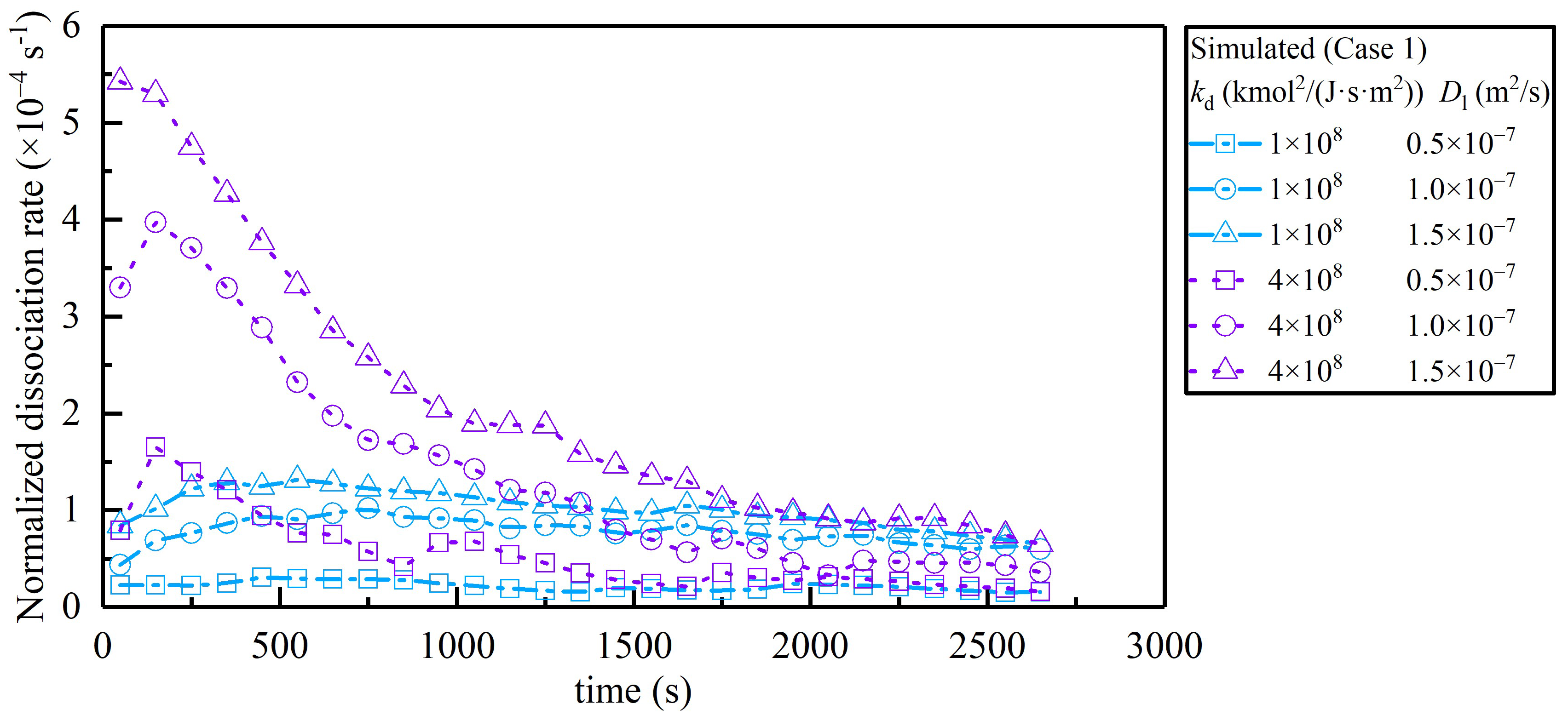
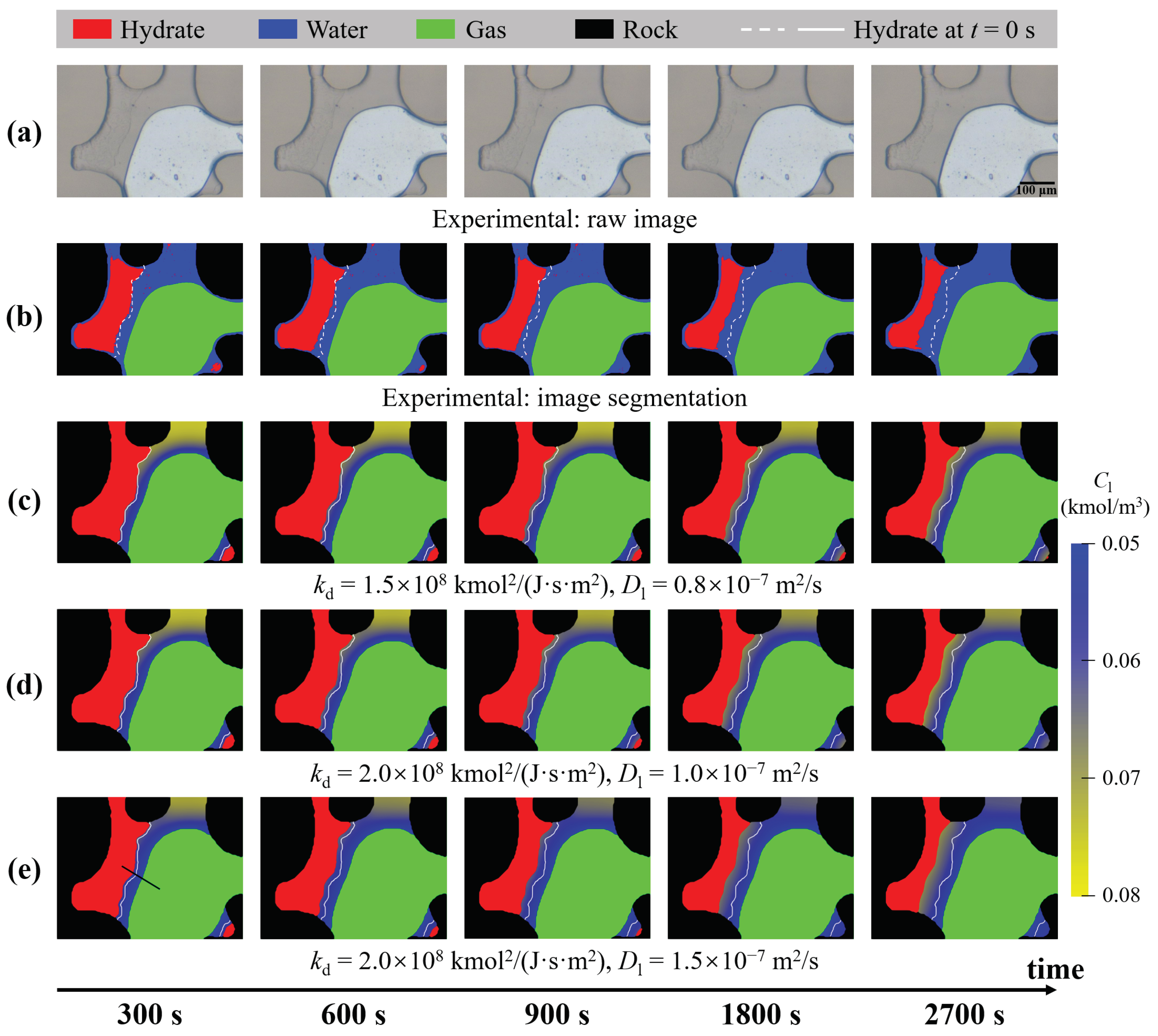
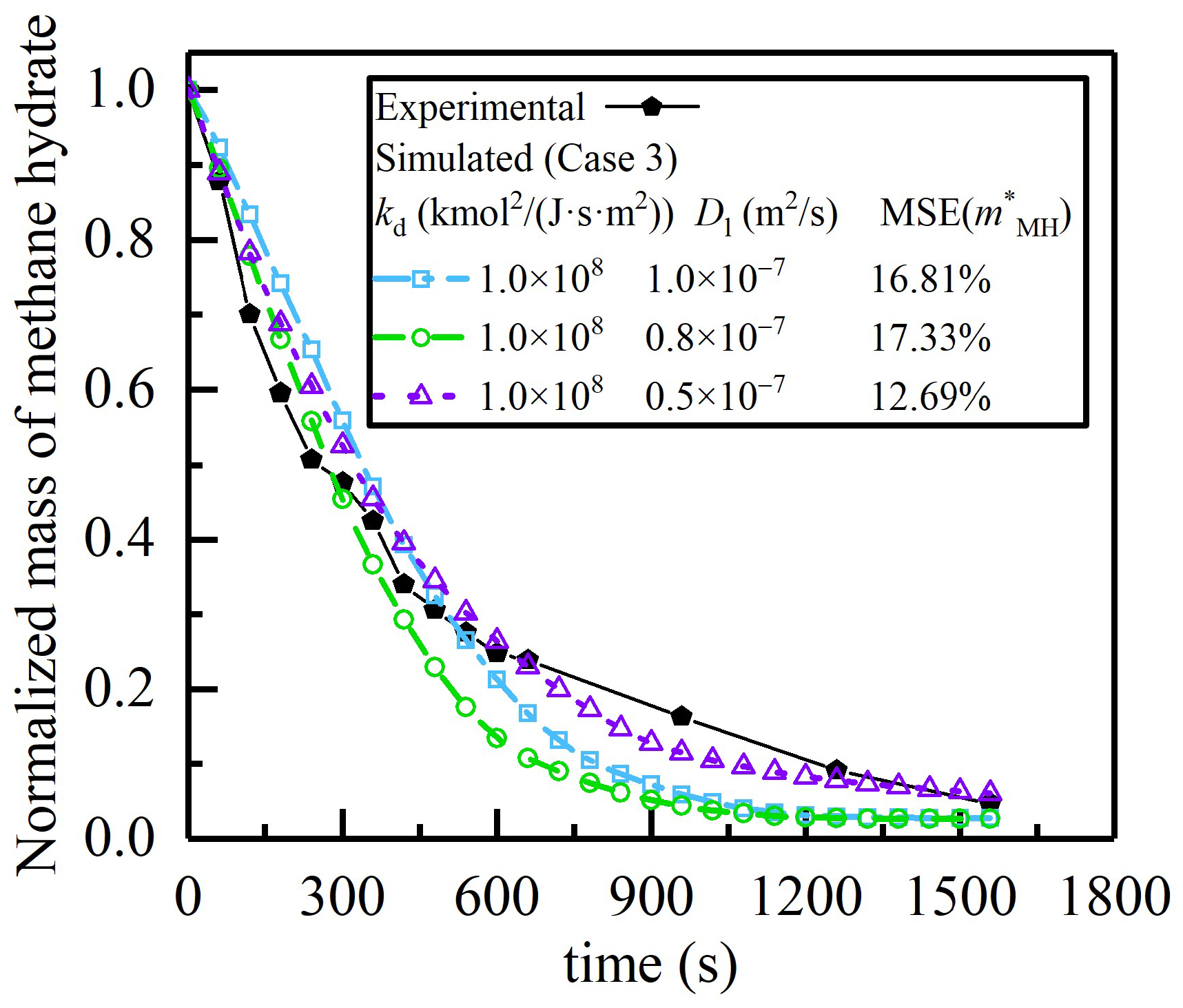
4.1. Estimation of Kinetic Parameters
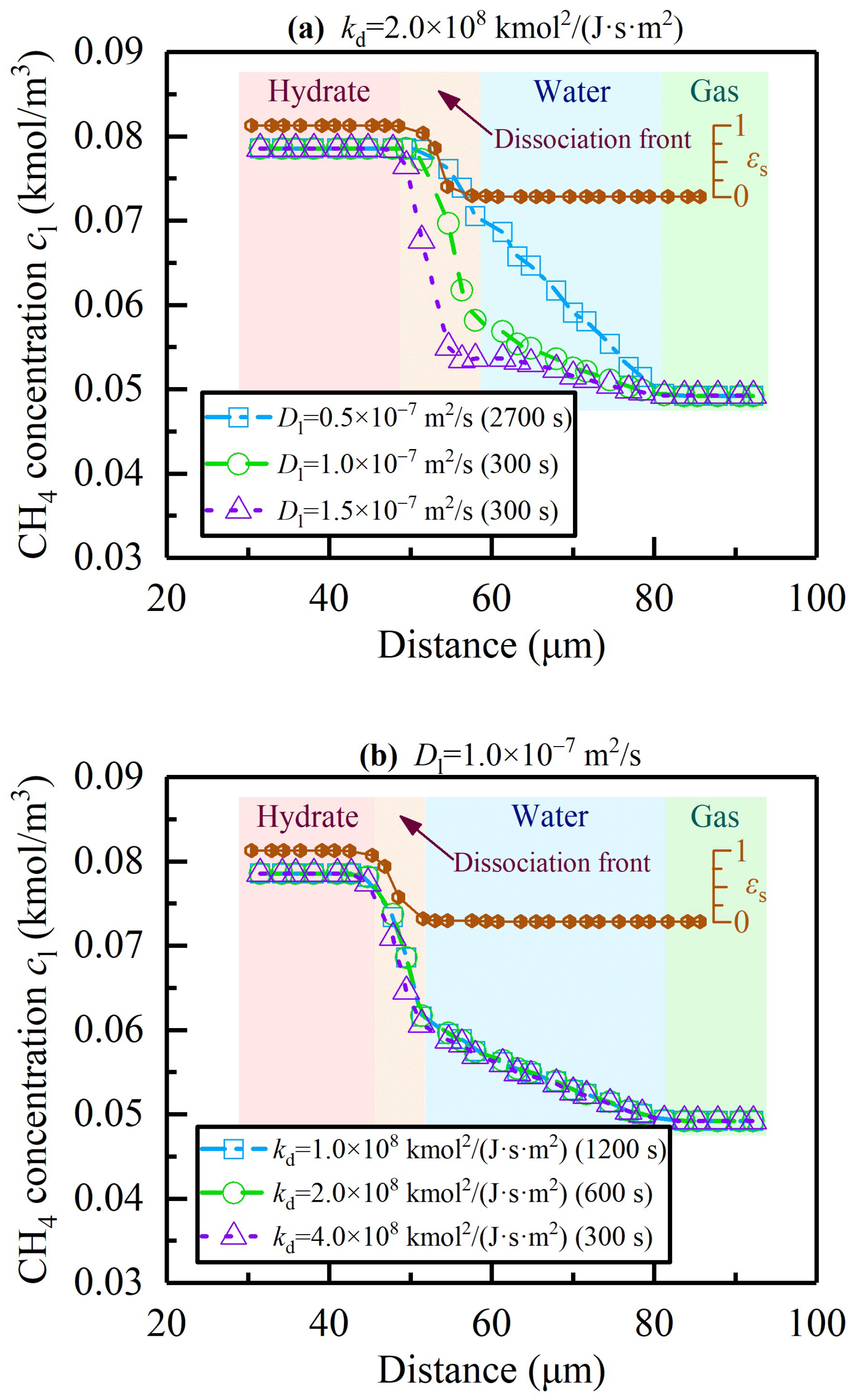
4.2. Verification of Unresolved Darcy-Scale Simulations
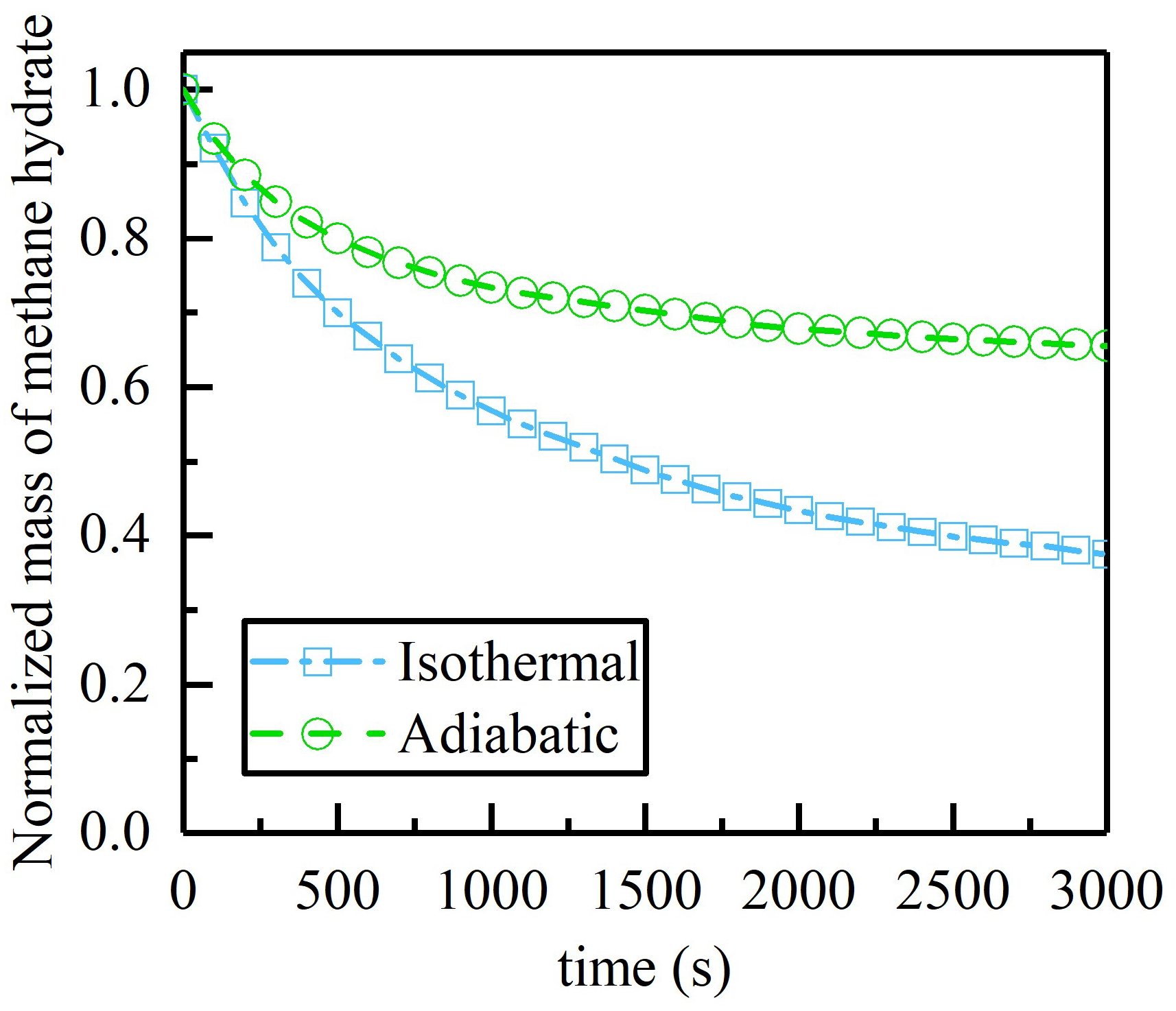
4.3. Effect of Temperature on Hydrate Dissociation
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Nomenclature
| Single-field concentration, kmol·m−3 | |
| Cp | Single-field heat capacity, kJ·kg−1·K−1 |
| Cp,s | Heat capacity of solid, kJ·kg−1·K−1 |
| Cp,f | Heat capacity of fluid, kJ·kg−1·K−1 |
| Cp,l | Heat capacity of water, kJ·kg−1·K−1 |
| Cp,g | Heat capacity of methane gas, kJ·kg−1·K−1 |
| Effective diffusion coefficien, m2·s−1 | |
| Dg | Diffusion coefficient of methane in gas phase, m2·s−1 |
| Dl | Diffusion coefficient of methane in liquid phase, m2·s−1 |
| Activation energy of methane hydrate dissociation, K | |
| Surface tension force, kg·m·s−2 | |
| Dissociation flux of methane hydrate, kmol·s−1·m−2 | |
| Rate constant of methane hydrate dissociation, kmol2·J−1·s−1·m−2 | |
| Ha | Henry’s constant |
| Standard enthalpy of methane hydrate dissociation, kJ·kg−1 | |
| Molar mass of methane hydrate, kg·kmol−1 | |
| Molar mass of methane gas, kg·kmol−1 | |
| Molar mass of water, kg·kmol−1 | |
| Hydrate number | |
| Source term in concentration equation, kmol·s−1·m−3 | |
| Normal vector of gas–liquid interface | |
| Single-field pressure, MPa | |
| Capillary pressure, MPa | |
| R | Ideal gas constant, 8.314 J·mol−1·K−1 |
| SH | Source term in energy equation, J·m−3 |
| Single-field temperature, K | |
| T | Dissociation temperature, K |
| Single-field velocity, m·s−1 | |
| Solubility of methane in an aqueous solution that is hypothetically in equilibrium with the hydrate phase | |
| Local solubility of methane in an aqueous solution | |
| Greek letters | |
| Volume fraction of gas phase | |
| Volume fraction of liquid phase | |
| Volume fraction of fluid phase, namely, porosity | |
| Volume fraction of solid phase, | |
| Single-field thermal conductivity, W·m−1·K−1 | |
| Thermal conductivity of solid phase, W·m−1·K−1 | |
| Thermal conductivity of methane gas, W·m−1·K−1 | |
| Thermal conductivity of water, W·m−1·K−1 | |
| Single-field dynamic viscosity, N·s·m−2 | |
| Dynamic viscosity of gas phase, N·s·m−2 | |
| Dynamic viscosity of liquid phase, N·s·m−2 | |
| Contact angle, ° | |
| Single-field density, kg·m−3 | |
| Density of gas phase, kg·m−3 | |
| Density of liquid phase, kg·m−3 | |
| Density of solid phase, kg·m−3 | |
Appendix A. Details of Model Parameters and Source Terms
Appendix A.1. Source Terms
Appendix A.2. Parameters in Momentum Equation
Appendix A.3. Parameters in Concentration Equation
References
- Sloan, E.D. Gas hydrates: Review of physical/chemical properties. Energy Fuels 1998, 12, 191–196. [Google Scholar] [CrossRef]
- Klauda, J.B.; Sandler, S.I. Global distribution of methane hydrate in ocean sediment. Energy Fuels 2005, 19, 459–470. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Holder, G.D. Methane hydrates potential as a future energy source. Fuel Process. Technol. 2001, 71, 181–186. [Google Scholar] [CrossRef]
- Song, Y.; Yang, L.; Zhao, J.; Liu, W.; Yang, M.; Li, Y.; Liu, Y.; Li, Q. The status of natural gas hydrate research in China: A review. Renew. Sustain. Energy Rev. 2014, 31, 778–791. [Google Scholar] [CrossRef]
- Hassanpouryouzband, A.; Joonaki, E.; Farahani, M.V.; Takeya, S.; Ruppel, C.; Yang, J.; English, N.J.; Schicks, J.M.; Edlmann, K.; Mehrabian, H. Gas hydrates in sustainable chemistry. Chem. Soc. Rev. 2020, 49, 5225–5309. [Google Scholar] [CrossRef]
- Sloan Jr, E.D. Fundamental principles and applications of natural gas hydrates. Nature 2003, 426, 353–359. [Google Scholar] [CrossRef]
- Pang, W.; Xu, W.; Sun, C.; Zhang, C.; Chen, G. Methane hydrate dissociation experiment in a middle-sized quiescent reactor using thermal method. Fuel 2009, 88, 497–503. [Google Scholar] [CrossRef]
- Ji, C.; Ahmadi, G.; Smith, D.H. Natural gas production from hydrate decomposition by depressurization. Chem. Eng. Sci. 2001, 56, 5801–5814. [Google Scholar] [CrossRef]
- Yuan, Q.; Sun, C.-Y.; Yang, X.; Ma, P.-C.; Ma, Z.-W.; Li, Q.-P.; Chen, G.-J. Gas production from methane-hydrate-bearing sands by ethylene glycol injection using a three-dimensional reactor. Energy Fuels 2011, 25, 3108–3115. [Google Scholar] [CrossRef]
- Yang, J.; Dai, X.; Xu, Q.; Liu, Z.; Zan, C.; Long, W.; Shi, L. Pore-scale study of multicomponent multiphase heat and mass transfer mechanism during methane hydrate dissociation process. Chem. Eng. J. 2021, 423, 130206. [Google Scholar] [CrossRef]
- Anderson, B.J.; Kurihara, M.; White, M.D.; Moridis, G.J.; Wilson, S.J.; Pooladi-Darvish, M.; Gaddipati, M.; Masuda, Y.; Collett, T.S.; Hunter, R.B. Regional long-term production modeling from a single well test, Mount Elbert gas hydrate stratigraphic test well, Alaska North slope. Mar. Pet. Geol. 2011, 28, 493–501. [Google Scholar] [CrossRef]
- Farahani, M.V.; Guo, X.; Zhang, L.; Yang, M.; Hassanpouryouzband, A.; Zhao, J.; Yang, J.; Song, Y.; Tohidi, B. Effect of thermal formation/dissociation cycles on the kinetics of formation and pore-scale distribution of methane hydrates in porous media: A magnetic resonance imaging study. Sustain. Energy Fuels 2021, 5, 1567–1583. [Google Scholar] [CrossRef]
- Li, X.-Y.; Li, X.-S.; Wang, Y.; Li, G.; Zhang, Y.; Hu, H.-Q.; Wan, K.; Zeng, H.-P. Influence of particle size on the heat and mass transfer characteristics of methane hydrate formation and decomposition in porous media. Energy Fuels 2021, 35, 2153–2164. [Google Scholar] [CrossRef]
- Yin, Z.; Moridis, G.; Tan, H.K.; Linga, P. Numerical analysis of experimental studies of methane hydrate formation in a sandy porous medium. Appl. Energy 2018, 220, 681–704. [Google Scholar] [CrossRef]
- Yu, M.; Li, W.; Jiang, L.; Wang, X.; Yang, M.; Song, Y. Numerical study of gas production from methane hydrate deposits by depressurization at 274 K. Appl. Energy 2018, 227, 28–37. [Google Scholar] [CrossRef]
- Lei, L.; Seol, Y.; Jarvis, K. Pore-scale visualization of methane hydrate-bearing sediments with micro-CT. Geophys. Res. Lett. 2018, 45, 5417–5426. [Google Scholar] [CrossRef]
- He, G.; Luo, X.; Zhang, H.; Bi, J. Pore-scale identification of multi-phase spatial distribution of hydrate bearing sediment. J. Geophys. Eng. 2018, 15, 2310–2317. [Google Scholar] [CrossRef]
- Tohidi, B.; Anderson, R.; Clennell, M.B.; Burgass, R.W.; Biderkab, A.B. Visual observation of gas-hydrate formation and dissociation in synthetic porous media by means of glass micromodels. Geology 2001, 29, 867–870. [Google Scholar] [CrossRef]
- Katsuki, D.; Ohmura, R.; Ebinuma, T.; Narita, H. Methane hydrate crystal growth in a porous medium filled with methane-saturated liquid water. Philos. Mag. 2007, 87, 1057–1069. [Google Scholar] [CrossRef]
- Almenningen, S.; Flatlandsmo, J.; Fernø, M.A.; Ersland, G. Multiscale laboratory verification of depressurization for production of sedimentary methane hydrates. SPE J. 2017, 22, 138–147. [Google Scholar] [CrossRef]
- Farahani, M.V.; Hassanpouryouzband, A.; Yang, J.; Tohidi, B. Development of a coupled geophysical–geothermal scheme for quantification of hydrates in gas hydrate-bearing permafrost sediments. PCCP 2021, 23, 24249–24264. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.-M.; Hsieh, L.-H.C.; Huang, C.-Y.; Sean, W.-Y. Direct numerical simulation of CO2 hydrate dissociation in pore-scale flow by applying CFD method. Int. J. Heat Mass Transf. 2017, 107, 300–306. [Google Scholar] [CrossRef]
- Mohammadmoradi, P.; Kantzas, A. Direct geometrical simulation of pore space evolution through hydrate dissociation in methane hydrate reservoirs. Mar. Pet. Geol. 2018, 89, 786–798. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, C.; Zhang, K.; Zhang, L.; Yao, J.; Sun, H.; Yang, Y. Pore-scale investigation of methane hydrate dissociation using the lattice Boltzmann method. Water Resour. Res. 2019, 55, 8422–8444. [Google Scholar] [CrossRef]
- Wang, X.; Dong, B.; Chen, C.; Li, W.; Song, Y. Pore-scale investigation on the influences of mass-transfer-limitation on methane hydrate dissociation using depressurization. Int. J. Heat Mass Transf. 2019, 144, 118656. [Google Scholar] [CrossRef]
- Wang, X.; Dong, B.; Wang, F.; Li, W.; Song, Y. Pore-scale investigations on the effects of ice formation/melting on methane hydrate dissociation using depressurization. Int. J. Heat Mass Transf. 2019, 131, 737–749. [Google Scholar] [CrossRef]
- Yang, J.; Xu, Q.; Liu, Z.; Shi, L. Pore-scale study of the multiphase methane hydrate dissociation dynamics and mechanisms in the sediment. Chem. Eng. J. 2022, 430, 132786. [Google Scholar] [CrossRef]
- Soulaine, C.; Roman, S.; Kovscek, A.; Tchelepi, H.A. Mineral dissolution and wormholing from a pore-scale perspective. J. Fluid Mech. 2017, 827, 457–483. [Google Scholar] [CrossRef]
- Scheibe, T.D.; Perkins, W.A.; Richmond, M.C.; McKinley, M.I.; Romero-Gomez, P.D.; Oostrom, M.; Wietsma, T.W.; Serkowski, J.A.; Zachara, J.M. Pore-scale and multiscale numerical simulation of flow and transport in a laboratory-scale column. Water Resour. Res. 2015, 51, 1023–1035. [Google Scholar] [CrossRef]
- Xu, Q.; Dai, X.; Yang, J.; Liu, Z.; Shi, L. Image-based modelling of coke combustion in a multiscale porous medium using a micro-continuum framework. J. Fluid Mech. 2022, 932, A51. [Google Scholar] [CrossRef]
- Yang, F.; Stack, A.G.; Starchenko, V. Micro-continuum approach for mineral precipitation. Sci. Rep. 2021, 11, 3495. [Google Scholar] [CrossRef] [PubMed]
- Horgue, P.; Prat, M.; Quintard, M. A penalization technique applied to the “Volume-Of-Fluid” method: Wettability condition on immersed boundaries. Comput. Fluids 2014, 100, 255–266. [Google Scholar] [CrossRef]
- Soulaine, C.; Roman, S.; Kovscek, A.; Tchelepi, H.A. Pore-scale modelling of multiphase reactive flow: Application to mineral dissolution with production of. J. Fluid Mech. 2018, 855, 616–645. [Google Scholar] [CrossRef]
- Carrillo, F.J.; Bourg, I.C.; Soulaine, C. Multiphase flow modeling in multiscale porous media: An open-source micro-continuum approach. J. Comput. Phys. X 2020, 8, 100073. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, J.; Xu, Q.; Shi, L. Improved micro-continuum approach for capillary-dominated multiphase flow with reduced spurious velocity. Phys. Fluids 2022, 34, 122108. [Google Scholar] [CrossRef]
- Sean, W.Y.; Sato, T.; Yamasaki, A.; Kiyono, F. CFD and experimental study on methane hydrate dissociation. Part II. General cases. AlChE J. 2007, 53, 2148–2160. [Google Scholar] [CrossRef]
- Maes, J.; Menke, H.P. GeoChemFoam: Direct modelling of multiphase reactive transport in real pore geometries with equilibrium reactions. Transp. Porous Media 2021, 139, 271–299. [Google Scholar] [CrossRef]
- Jasak, H.; Jemcov, A.; Tukovic, Z. OpenFOAM: A C++ library for complex physics simulations. In Proceedings of the International Workshop on Coupled Methods in Numerical Dynamics, Dubrovnik, Croatia, 19–21 September 2007; pp. 1–20. [Google Scholar]
- Van Leer, B. Towards the ultimate conservative difference scheme. J. Comput. Phys. 1997, 135, 229–248. [Google Scholar] [CrossRef]
- Maes, J.; Soulaine, C. A unified single-field Volume-of-Fluid-based formulation for multi-component interfacial transfer with local volume changes. J. Comput. Phys. 2020, 402, 109024. [Google Scholar] [CrossRef]
- Issa, R.I. Solution of the implicitly discretised fluid flow equations by operator-splitting. J. Comput. Phys. 1986, 62, 40–65. [Google Scholar] [CrossRef]
- Maes, J. GeoChemFoam User Guide. Available online: https://github.com/GeoChemFoam/GeoChemFoam/blob/main/GeoChemFoam-User-Guide.pdf (accessed on 9 March 2022).
- Yang, J. Pore-scale Visualization of Hydrate Dissociation and Mass Transfer during Depressurization Using Microfluidic Experiments. Fuel, 2023; in press. [Google Scholar]
- Brooks, R.H. Hydraulic Properties of Porous Media; Colorado State University: Fort Collins, CO, USA, 1965. [Google Scholar]
- Van Genuchten, M.T. A closed-form equation for predicting the hydraulic conductivity of unsaturated soils. Soil Sci. Soc. Am. J. 1980, 44, 892–898. [Google Scholar] [CrossRef]
- Xu, Q.; Long, W.; Jiang, H.; Ma, B.; Zan, C.; Ma, D.; Shi, L. Quantification of the microstructure, effective hydraulic radius and effective transport properties changed by the coke deposition during the crude oil in-situ combustion. Chem. Eng. J. 2018, 331, 856–869. [Google Scholar] [CrossRef]
- Haroun, Y.; Legendre, D.; Raynal, L. Volume of fluid method for interfacial reactive mass transfer: Application to stable liquid film. Chem. Eng. Sci. 2010, 65, 2896–2909. [Google Scholar] [CrossRef]
- Maes, J.; Soulaine, C. A new compressive scheme to simulate species transfer across fluid interfaces using the volume-of-fluid method. Chem. Eng. Sci. 2018, 190, 405–418. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Case 1 | Cases 2 and 3 | ||
|---|---|---|---|---|
| Right | Others | Bottom | Others | |
| Zero gradient | Zero gradient | Zero gradient | Zero gradient | |
| Pressure, | ||||
| Velocity, | ||||
| Concentration, | ||||
| Temperature, | ||||
| Category | Parameter | Value |
|---|---|---|
| Physical properties of methane | Density (kg/m3) | 20 |
| Kinematic viscosity (m2/s) | 10−5 | |
| Thermal conductivity (W/(m·K)) | 0.045 | |
| Specific heat capacity (kJ/(kg·K)) | 3.2 | |
| Effective diffusion coefficient in the liquid phase (m2/s) | (0.5~1.5) × 10−7 | |
| Diffusion coefficient in the gas phase (m2/s) | 1 × 10−5 | |
| Henry’s coefficient (-) | 0.05 | |
| Physical properties of water | Density (kg/m3) | 1000 |
| Kinematic viscosity (m2/s) | 10−6 | |
| Thermal conductivity (W/(m·K)) | 0.55 | |
| Specific heat capacity (kJ/(kg·K)) | 4.2 | |
| Physical properties of methane hydrate | Density (kg/m3) | 900 |
| Thermal conductivity (W/(m·K)) | 0.49 | |
| Specific heat capacity (kJ/(kg·K)) | 2.1 | |
| Kinetic parameters of hydrate dissociation | Rate constant (kmol2/(J·s·m2)) | (1~4) × 108 |
| Activation heat (K) | 9399 | |
| Dissociation heat (kJ/mol) | 51.86 |
| Mesh No. | Grid Number | Grid Size (μm) | ||||
|---|---|---|---|---|---|---|
| Coarse | 105 × 75 | 5.0 | 0.1277 | −4.06% | 0.0498 | −0.4% |
| Normal | 175 × 125 | 3.0 | 0.1321 | −0.75% | 0.0501 | 0.2% |
| Fine | 350 × 250 | 1.5 | 0.1329 | −0.15% | 0.0499 | −0.2% |
| Finer | 525 × 375 | 1.0 | 0.1331 | / | 0.0500 | 0.0% |
| Case Description | Case No. | kd (×108 kmol2/(J·s·m2)) | Dl (×10−7 m2/s) | |
|---|---|---|---|---|
| Case 1: Hydrate dissociation with a pressure difference of 0.8 MPa | 1a | 1.0 | 0.5 | 30.22% |
| 1b | 1.0 | 1.0 | 16.03% | |
| 1c | 1.0 | 1.5 | 9.50% | |
| 1d | 2.0 | 0.5 | 29.16% | |
| 1e | 2.0 | 1.0 | 4.08% | |
| 1f | 2.0 | 1.5 | 6.86% | |
| 1g | 4.0 | 0.5 | 20.37% | |
| 1h | 4.0 | 1.0 | 8.62% | |
| 1i | 4.0 | 1.5 | 26.31% | |
| 1j | 1.5 | 0.8 | 12.39% |
| Case Description | Case No. | kd (×108 kmol2/(J·s·m2)) | Dl (×10−7 m2/s) | |
|---|---|---|---|---|
| Case 2: Hydrate dissociation with pressure difference of 2.84 MPa | 2a | 0.5 | 0.5 | 103.54% |
| 2b | 0.5 | 1.0 | 75.74% | |
| 2c | 0.5 | 1.5 | 50.97% | |
| 2d | 1.0 | 0.5 | 59.72% | |
| 2e | 1.0 | 1.0 | 15.05% | |
| 2f | 1.0 | 1.5 | 33.59% | |
| 2g | 2.0 | 0.5 | 7.50% | |
| 2h | 2.0 | 1.0 | 53.13% | |
| 2i | 2.0 | 1.5 | 67.01% | |
| 2j | 1.5 | 0.8 | 19.82% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Xu, Q.; Yang, J.; Shi, L. Pore-Scale Modeling of Methane Hydrate Dissociation Using a Multiphase Micro-Continuum Framework. Energies 2023, 16, 7599. https://doi.org/10.3390/en16227599
Liu Z, Xu Q, Yang J, Shi L. Pore-Scale Modeling of Methane Hydrate Dissociation Using a Multiphase Micro-Continuum Framework. Energies. 2023; 16(22):7599. https://doi.org/10.3390/en16227599
Chicago/Turabian StyleLiu, Zhiying, Qianghui Xu, Junyu Yang, and Lin Shi. 2023. "Pore-Scale Modeling of Methane Hydrate Dissociation Using a Multiphase Micro-Continuum Framework" Energies 16, no. 22: 7599. https://doi.org/10.3390/en16227599