
A Comprehensive Review of Defects Genealogy in Olivines: From the Mineral World to Modern Electrode Materials


Abstract
:1. Introduction
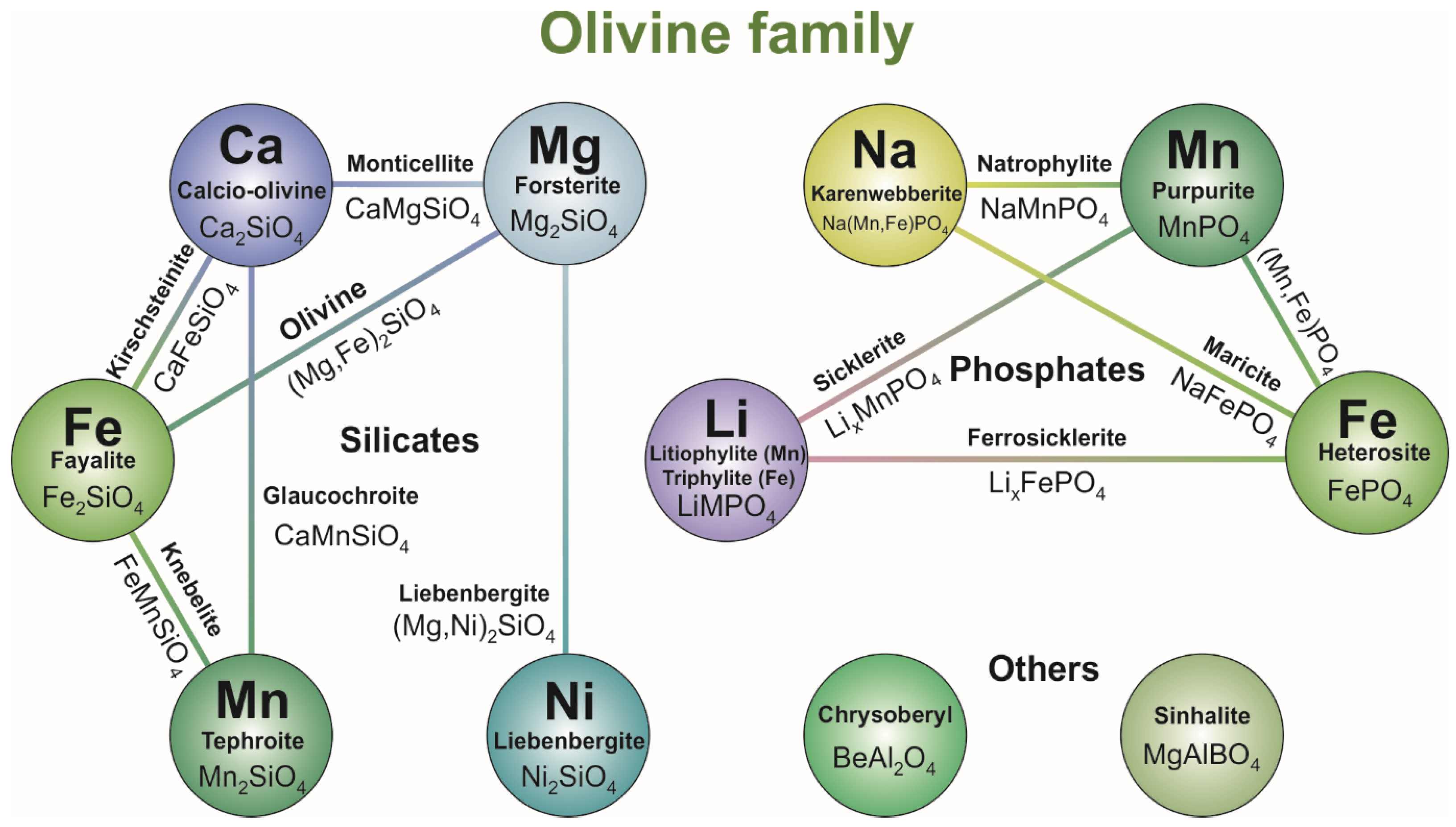
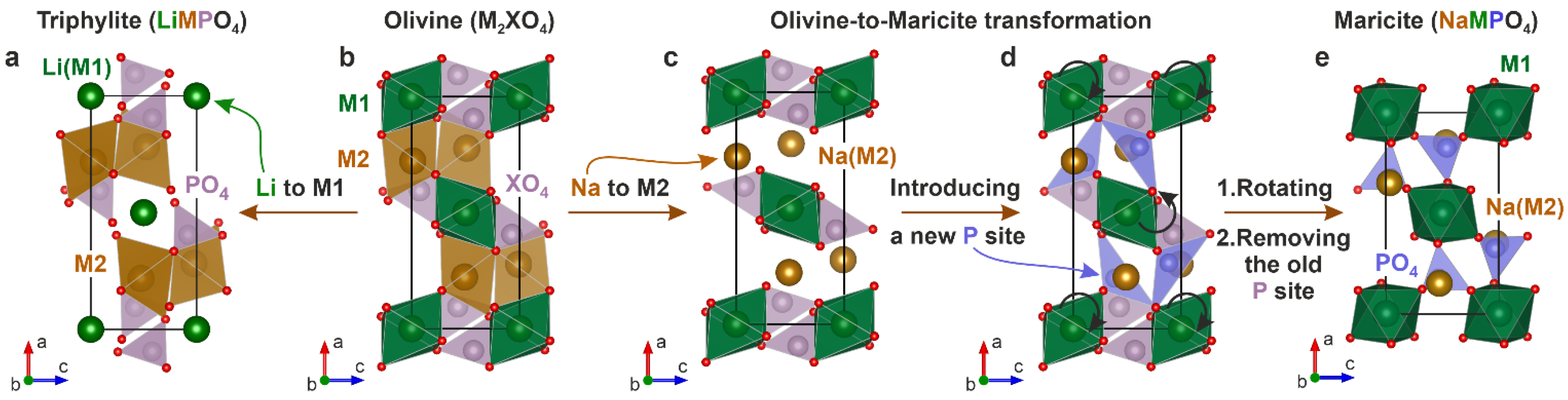
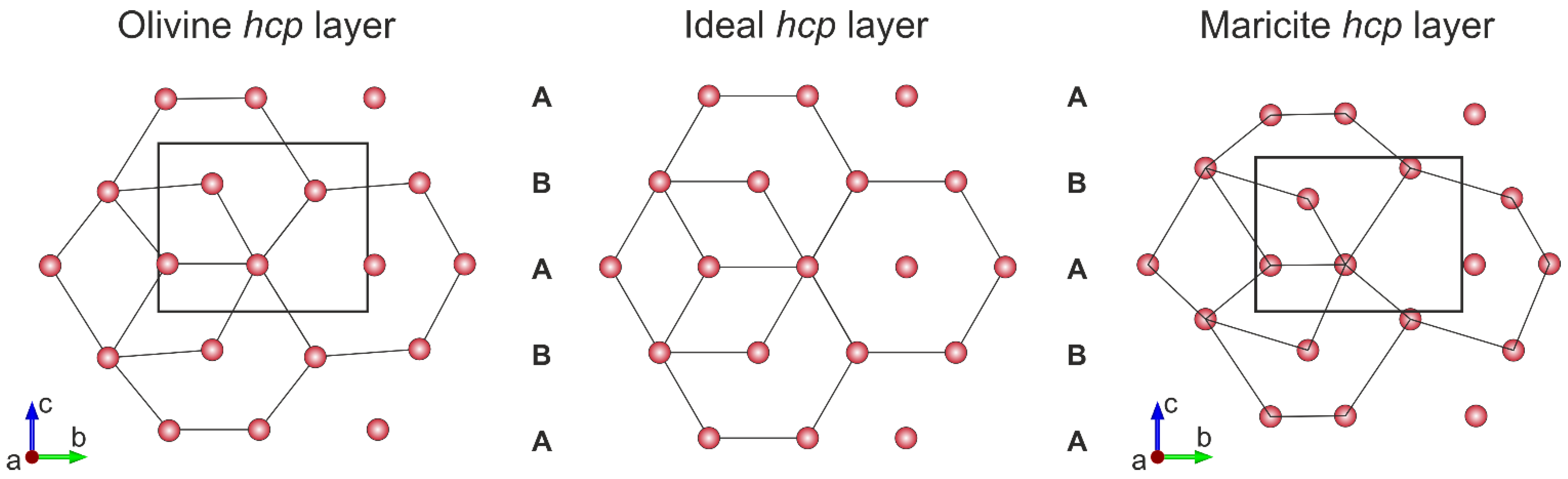
2. Crystal Structure, Stability and Variety of Olivine-Type Minerals
3. Point Defects in Cationic Sublattice: The Influence on Transport and Electrochemical Properties of Triphylite Cathodes
3.1. General Overview of Point Defects
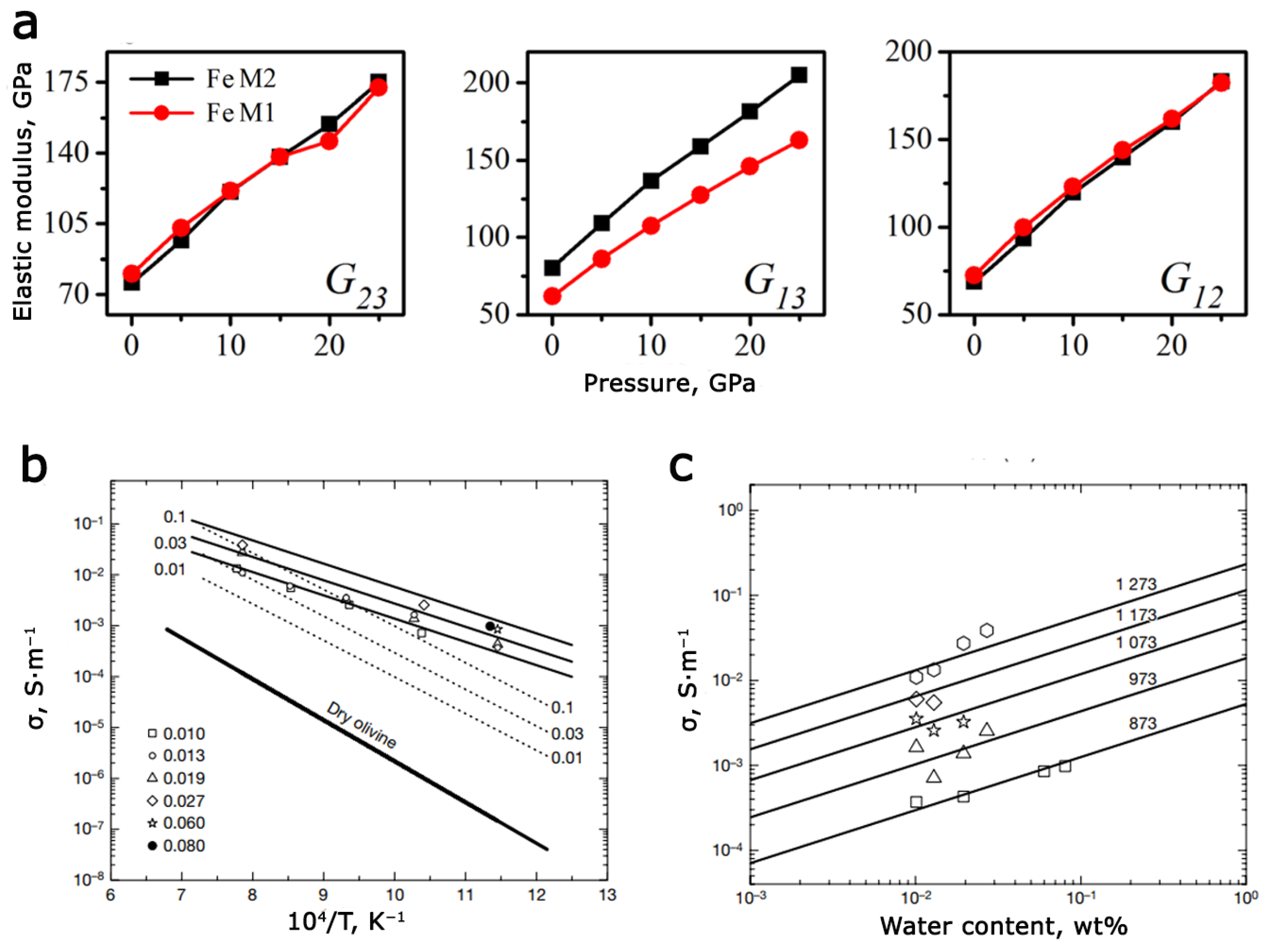
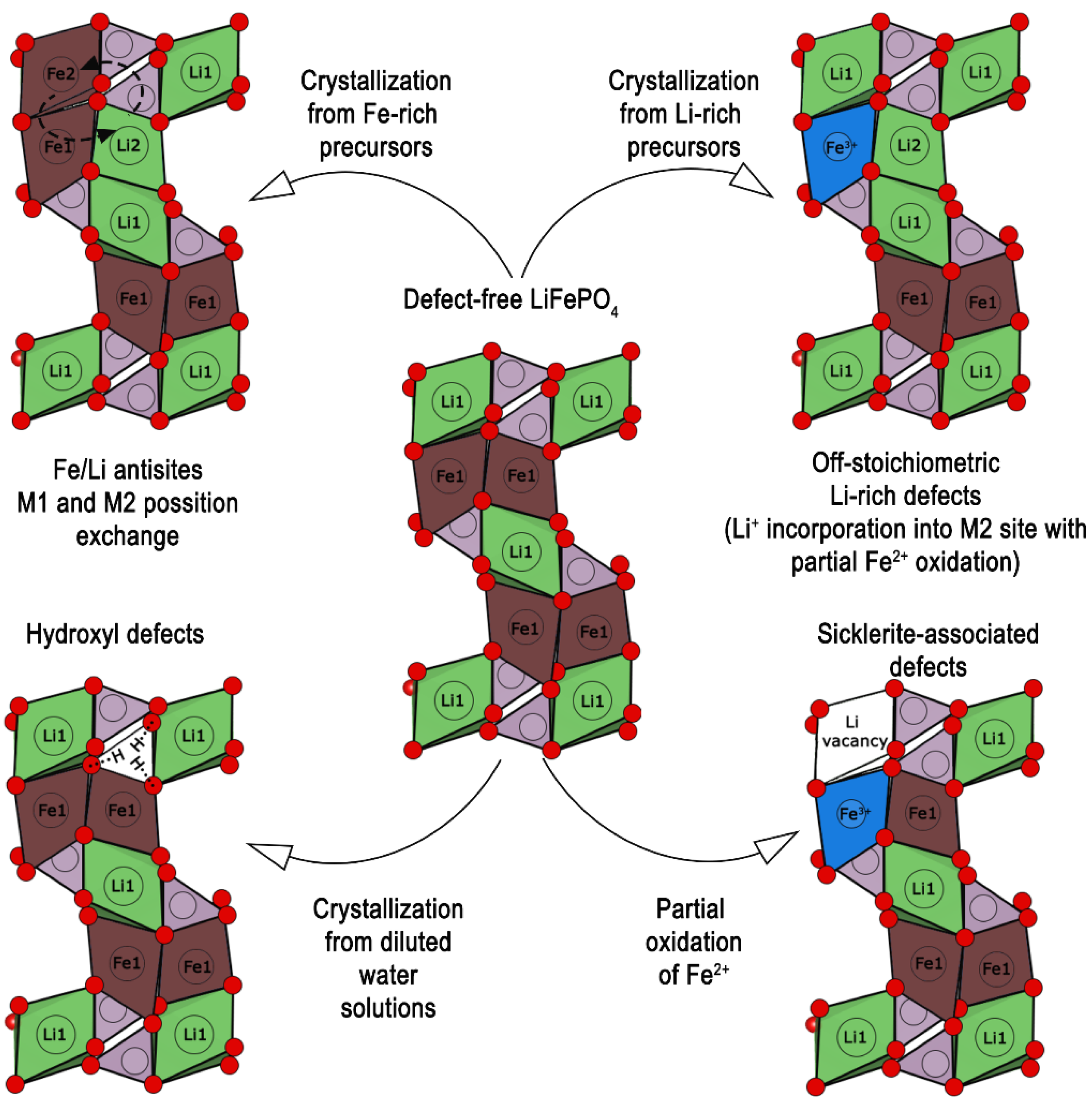
3.2. Defects of Cationic and Anionic Sublattice in Olivine Minerals
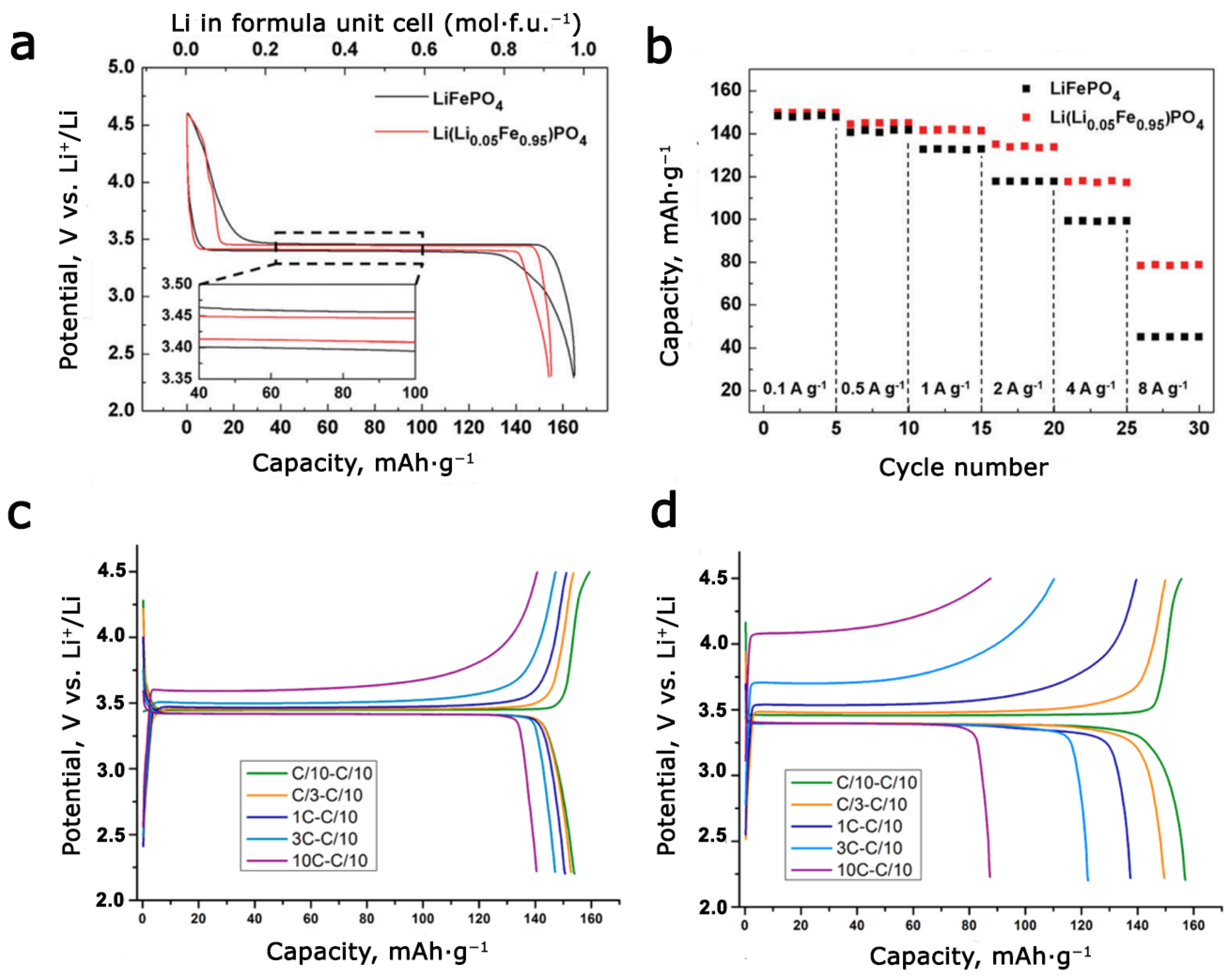
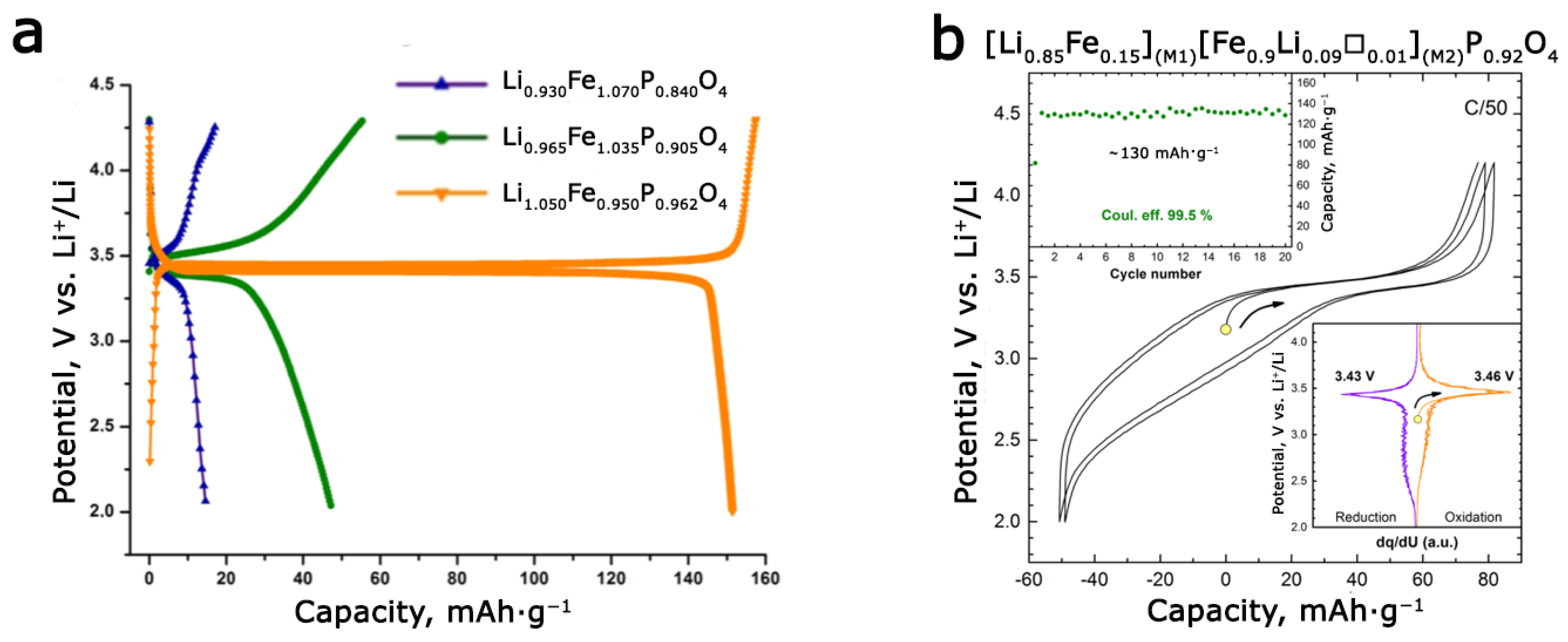
3.3. Defects of Cationic and Anionic Sublattice in Synthetic and Natural Triphylites
4. Discussion and Outlook
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (Mg,Fe)2SiO4 | LiFePO4 | Description |
| Vacant Fe (M2) site | ||
| Vacant Mg/Li (M1) site | ||
| Vacant O site | ||
| Vacant Si/P site | ||
| A proton at an unoccupied interstitial position | ||
| A proton in the Mg/Li site | ||
| A proton in the Fe site | ||
| Mg2+/Li+ at an unoccupied interstitial position | ||
| Fe2+ at an unoccupied interstitial position | ||
| O2− at an unoccupied interstitial position | ||
| Si4+ at an unoccupied interstitial position | ||
| e′ | e′ | Electron in the conduction band |
| h· | h· | Hole in the valence band |
| Fe3+ in the Mg/Li (M1) site (antisite defect) | ||
| Fe3+ in the Fe (M2) site (Fe oxidation) | ||
| Mg2+/Li+ ion in the Mg/Li site | ||
| Fe2+ ion in the Mg/Li site (antisite defect) | ||
| Fe2+ ion in the Fe site | ||
| Si4+/P5+ ion in the Si/P site | ||
| O2− ion in the O site |
References
- Berry, A.J.; Hermann, J.; O’Neill, H.S.C.; Foran, G.J. Fingerprinting the Water Site in Mantle Olivine. Geology 2005, 33, 869. [Google Scholar] [CrossRef]
- The Effect of Water on the Electrical Conductivity of Olivine|Nature. Available online: https://www.nature.com/articles/nature05256 (accessed on 28 March 2023).
- Water in Earth’s Mantle: The Role of Nominally Anhydrous Minerals | Science. Available online: https://www.science.org/doi/10.1126/science.255.5050.1391 (accessed on 28 March 2023).
- Padhi, A.K.; Nanjundaswamy, K.S.; Goodenough, J.B. Phospho-olivines as Positive-Electrode Materials for Rechargeable Lithium Batteries. J. Electrochem. Soc. 1997, 144, 1188. [Google Scholar] [CrossRef]
- Zhang, S.S. Problems and Their Origins of Ni-Rich Layered Oxide Cathode Materials. Energy Storage Mater. 2020, 24, 247–254. [Google Scholar] [CrossRef]
- Julien, C.M.; Mauger, A.; Zaghib, K.; Groult, H. Comparative Issues of Cathode Materials for Li-Ion Batteries. Inorganics 2014, 2, 132–154. [Google Scholar] [CrossRef] [Green Version]
- Nazarov, E.E.; Dembitskiy, A.D.; Trussov, I.A.; Tyablikov, O.A.; Glazkova, I.S.; Alexey, S.V.; Presniakov, I.A.; Mikheev, I.V.; Morozov, A.V.; Nikitina, V.A.; et al. A Li-Rich Strategy towards Advanced Mn-Doped Triphylite Cathodes for Li-Ion Batteries. Energy Adv. 2023, 2, 328–337. [Google Scholar] [CrossRef]
- Nazarov, E.E.; Tyablikov, O.A.; Nikitina, V.A.; Antipov, E.V.; Fedotov, S.S. Polyacrylonitrile-Derived Carbon Nanocoating for Long-Life High-Power Phosphate Electrodes. Appl. Nano 2023, 4, 25–37. [Google Scholar] [CrossRef]
- Drozhzhin, O.A.; Sobolev, A.V.; Sumanov, V.D.; Glazkova, I.S.; Aksyonov, D.A.; Grebenshchikova, A.D.; Tyablikov, O.A.; Alekseeva, A.M.; Mikheev, I.V.; Dovgaliuk, I.; et al. Exploring the Origin of the Superior Electrochemical Performance of Hydrothermally Prepared Li-Rich Lithium Iron Phosphate Li1+δFe1−δPO4. J. Phys. Chem. C 2020, 124, 126–134. [Google Scholar] [CrossRef]
- Ravnsbæk, D.B.; Xiang, K.; Xing, W.; Borkiewicz, O.J.; Wiaderek, K.M.; Gionet, P.; Chapman, K.W.; Chupas, P.J.; Chiang, Y.-M. Extended Solid Solutions and Coherent Transformations in Nanoscale Olivine Cathodes. Nano Lett. 2014, 14, 1484–1491. [Google Scholar] [CrossRef]
- Koleva, V.G.; Boyadzhieva, T.J.; Stoyanova, R.K. Crystal and Morphology Design of Dittmarite-Type Ammonium Iron–Manganese Phosphates, NH4Mn1–XFexPO4·H2O, as Precursors for Phospho-Olivine Electrodes. Cryst. Growth Des. 2019, 19, 3744–3754. [Google Scholar] [CrossRef]
- Freeze Drying under Vacuum Assisted Synthesis of LiFePO4@MWCNTs Composite with Phytic Acid as Phosphorus Source for Advanced Li-Storage-Science Direct. Available online: https://www.sciencedirect.com/science/article/abs/pii/S0042207X21004905 (accessed on 28 April 2023).
- Vignola, P.; Hatert, F.; Fransolet, A.-M.; Medenbach, O.; Diella, V.; Andò, S. Karenwebberite, Na(Fe2+,Mn2+)PO4, a New Member of the Triphylite Group from the Malpensata Pegmatite, Lecco Province, Italy. Am. Mineral. 2013, 98, 767–772. [Google Scholar] [CrossRef]
- Heubner, C.; Heiden, S.; Schneider, M.; Michaelis, A. In-Situ Preparation and Electrochemical Characterization of Submicron Sized NaFePO4 Cathode Material for Sodium-Ion Batteries. Electrochim. Acta 2017, 233, 78–84. [Google Scholar] [CrossRef]
- Zhu, Y.; Xu, Y.; Liu, Y.; Luo, C.; Wang, C. Comparison of Electrochemical Performances of Olivine NaFePO4 in Sodium-Ion Batteries and Olivine LiFePO4 in Lithium-Ion Batteries. Nanoscale 2012, 5, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Sumanov, V.D.; Aksyonov, D.A.; Drozhzhin, O.A.; Presniakov, I.; Sobolev, A.V.; Glazkova, I.; Tsirlin, A.A.; Rupasov, D.; Senyshyn, A.; Kolesnik, I.V.; et al. “Hydrotriphylites” Li1–xFe1+x(PO4)1–y(OH)4y as Cathode Materials for Li-Ion Batteries. Chem. Mater. 2019, 31, 5035–5046. [Google Scholar] [CrossRef]
- Aksyonov, D.A.; Varlamova, I.; Trussov, I.A.; Savina, A.A.; Senyshyn, A.; Stevenson, K.J.; Abakumov, A.M.; Zhugayevych, A.; Fedotov, S.S. Hydroxyl Defects in LiFePO4 Cathode Material: DFT+ U and an Experimental Study. Inorg. Chem. 2021, 60, 5497–5506. [Google Scholar] [CrossRef] [PubMed]
- Burns, R.G. Site Preferences of Transition Metal Ions in Silicate Crystal Structures. Chem. Geol. 1970, 5, 275–283. [Google Scholar] [CrossRef]
- Brrle, J.D. Crystal structures of natural olivines. Am. Mineral. 1968, 53, 807–824. [Google Scholar]
- Aikawa, N.; Tokonami, M. Crystal Structure and Cation Distribution of “Cleavable” Olivines. Mineral. J. 1987, 13, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Avdeev, M.; Mohamed, Z.; Ling, C.D.; Lu, J.; Tamaru, M.; Yamada, A.; Barpanda, P. Magnetic Structures of NaFePO4 Maricite and Triphylite Polymorphs for Sodium-Ion Batteries. Inorg. Chem. 2013, 52, 8685–8693. [Google Scholar] [CrossRef]
- Stewart, D.B. Petrogenesis of Lithium-Rich Pegmatites. Am. Mineral. 1978, 63, 970–980. [Google Scholar]
- The Classification of Granitic Pegmatites Revisited | The Canadian Mineralogist | GeoScienceWorld. Available online: https://pubs.geoscienceworld.org/canmin/article-abstract/43/6/2005/126681/THE-CLASSIFICATION-OF-GRANITIC-PEGMATITES?redirectedFrom=fulltext (accessed on 28 March 2023).
- Li, Z.; Shinno, I. Next Nearest Neighbor Effects in Triphylite and Related Phosphate Minerals. Mineral. J. 1997, 19, 99–107. [Google Scholar] [CrossRef] [Green Version]
- The Crystal Structure of the New Mineral Maricite, NaFePO4 | Semantic Scholar. Available online: https://www.semanticscholar.org/paper/The-crystal-structure-of-the-new-mineral-maricite%2C-Page-Donnay/a440fc91063a0b9e8c0be502df61aef8c592076a (accessed on 28 March 2023).
- The Crystal Structure of NaMnPO4 | Semantic Scholar. Available online: https://www.semanticscholar.org/paper/The-crystal-structure-of-NaMnPO4-Moring-Kostiner/7b749288d2fce21372458ad07dba97d9389813dd (accessed on 28 March 2023).
- Rousse, G.; Rodriguez-Carvajal, J.; Patoux, S.; Masquelier, C. Magnetic Structures of the Triphylite LiFePO4 and of Its Delithiated Form FePO4. Chem. Mater. 2003, 15, 4082–4090. [Google Scholar] [CrossRef]
- Topochemical Synthesis of Sodium Metal Phosphate Olivines for Sodium-Ion Batteries | Chemistry of Materials. Available online: https://pubs.acs.org/doi/10.1021/cm200450y (accessed on 28 March 2023).
- Storage Performance of Mg+++ Substituted NaMnPO4 with an Olivine Structure-RSC Advances (RSC Publishing). Available online: https://pubs.rsc.org/en/content/articlelanding/2020/ra/d0ra05698g (accessed on 20 May 2023).
- Precursor-Based Methods for Low-Temperature Synthesis of Defectless NaMnPO4 with an Olivine- and Maricite-Type Structure-CrystEngComm (RSC Publishing). Available online: https://pubs.rsc.org/en/content/articlelanding/2013/ce/c3ce41545g (accessed on 20 May 2023).
- Denisov, I.P.; Yakovlev, V.Y. Formation of Self-Trapped Exitons and Defects in Alkali Halides under Pulsed Electron Irradiation. Izv. Akad. Nauk. Latv. SSR Seriya Fiz. I Tekhnicheskikh Nauk. 1990, 3, 66–72. [Google Scholar]
- H-Center and V-Center Defects in Hybrid Halide Perovskites | ACS Energy Letters. Available online: https://pubs.acs.org/doi/full/10.1021/acsenergylett.7b00995 (accessed on 28 March 2023).
- Chen, W.; Tegenkamp, C.; Pfnür, H.; Bredow, T. Color Centers in NaCl by Hybrid Functionals. Phys. Rev. B 2010, 82, 104106. [Google Scholar] [CrossRef] [Green Version]
- Gordon, R.B.; Nowick, A.S. Structure Sensitivity of the X-Ray Coloration of NaCl Crystals. Phys. Rev. 1956, 101, 977–983. [Google Scholar] [CrossRef]
- Polanco, C.A.; Lindsay, L. Thermal Conductivity of InN with Point Defects from First Principles. Phys. Rev. B 2018, 98, 014306. [Google Scholar] [CrossRef] [Green Version]
- Stern, R.; Wang, T.; Carrete, J.; Mingo, N.; Madsen, G.K.H. Influence of Point Defects on the Thermal Conductivity in FeSi. Phys. Rev. B 2018, 97, 195201. [Google Scholar] [CrossRef]
- Rissouli, K.; Benkhouja, K.; Ramos-Barrado, J.R.; Julien, C. Electrical Conductivity in Lithium Orthophosphates. Mater. Sci. Eng. B 2003, 98, 185–189. [Google Scholar] [CrossRef]
- Reznik, A.; Uzan-Saguy, C.; Kalish, R. Effects of Point Defects on the Electrical Properties of Doped Diamond. Diam. Relat. Mater. 2000, 9, 1051–1056. [Google Scholar] [CrossRef]
- Hong, J.; Wang, C.S.; Chen, X.; Upreti, S.; Whittingham, M.S. Vanadium Modified LiFePO4 Cathode for Li-Ion Batteries. Electrochem. Solid-State Lett. 2008, 12, A33. [Google Scholar] [CrossRef]
- Maier, J.; Amin, R. Defect Chemistry of LiFePO4. J. Electrochem. Soc. 2008, 155, A339. [Google Scholar] [CrossRef]
- Walker, A.M.; Woodley, S.M.; Slater, B.; Wright, K. A Computational Study of Magnesium Point Defects and Diffusion in Forsterite. Phys. Earth Planet. Inter. 2009, 172, 20–27. [Google Scholar] [CrossRef]
- Kröger, F.A.; Vink, H.J. Relations between the Concentrations of Imperfections in Crystalline Solids. In Solid State Physics; Seitz, F., Turnbull, D., Eds.; Academic Press: Cambridge, MA, USA, 1956; Volume 3, pp. 307–435. [Google Scholar]
- Zhang, B. An Overview of Fe–Mg Interdiffusion in Mantle Minerals. Surv. Geophys. 2017, 38, 727–755. [Google Scholar] [CrossRef]
- Ghose, S.; Wan, C. Strong site preference of Co2+ in olivine, Co1.10Mg0.90SiO4. Contrib. Miner. Pet. 1974, 47, 131–140. [Google Scholar] [CrossRef]
- Buening, D.K.; Buseck, P.R. Fe-Mg lattice diffusion in olivine. J. Geophys. Res. Atmos. 1973, 78, 6852–6862. [Google Scholar] [CrossRef]
- Misener, D.J. Cation Diffusion in Olivine to 1400 °C and 35 KB; The University of British Columbia: Kelowna, BC, USA, 1973. [Google Scholar]
- Chakraborty, S.; Knoche, R.; Schulze, H.; Rubie, D.C.; Dobson, D.; Ross, N.L.; Angel, R.J. Enhancement of Cation Diffusion Rates Across the 410-Kilometer Discontinuity in Earth’s Mantle. Science 1999, 283, 362–365. [Google Scholar] [CrossRef]
- Jaoul, O.; Bertran-Alvarez, Y.; Liebermann, R.C.; Price, G.D. Fe, Mg interdiffusion in olivine up to 9 GPa at T = 600–900 °C; experimental data and comparison with defect calculations. Phys. Earth Planet. Inter. 1995, 89, 199–218. [Google Scholar] [CrossRef]
- Dohmen, R.; Chakraborty, S. Fe–Mg Diffusion in Olivine II: Point Defect Chemistry, Change of Diffusion Mechanisms and a Model for Calculation of Diffusion Coefficients in Natural Olivine. Phys Chem Miner. 2007, 34, 409–430. [Google Scholar] [CrossRef]
- Tachibana, S.; Tamada, S.; Kawasaki, H.; Ozawa, K.; Nagahara, H. Interdiffusion of Mg–Fe in Olivine at 1400–1600 °C and 1 Atm Total Pressure. Phys. Chem. Miner. 2013, 40, 511–519. [Google Scholar] [CrossRef]
- Chakraborty, S. Diffusion Coefficients in Olivine, Wadsleyite and Ringwoodite. Rev. Mineral. Geochem. 2010, 72, 603–639. [Google Scholar] [CrossRef]
- Chatterjee, S.; Sengupta, S.; Saha-Dasgupta, T.; Chatterjee, K.; Mandal, N. Site Preference of Fe Atoms in FeMgSiO4 and FeMg(SiO3)2 Studied by Density Functional Calculations. Phys. Rev. B 2009, 79, 115103. [Google Scholar] [CrossRef]
- Redfern, S.A.T.; Artioli, G.; Rinaldi, R.; Henderson, C.M.B.; Knight, K.S.; Wood, B.J. Octahedral cation ordering in olivine at high temperature. II: An in situ neutron powder diffraction study on synthetic MgFeSiO4 (Fa50). Phys. Chem. Miner. 2000, 27, 630–637. [Google Scholar] [CrossRef]
- Kolesov, B.A.; Geiger, C.A. A Raman spectroscopic study of Fe–Mg olivines. Phys. Chem. Miner. 2004, 31, 142–154. [Google Scholar] [CrossRef]
- Das, P.K.; Mandal, N.; Arya, A. Effects of cation ordering on the elastic and electronic properties of Mg-Fe silicate phases at high pressures. J. Appl. Phys. 2017, 122, 225107. [Google Scholar] [CrossRef]
- Mei, S.; Kohlstedt, D.L. Influence of water on plastic deformation of olivine aggregates: 1. Diffusion creep regime. J. Geophys. Res. Solid Earth 2000, 105, 21457–21469. [Google Scholar] [CrossRef]
- Walker, A.M.; Hermann, J.; Berry, A.J.; O’Neill, H.S.C. Three Water Sites in Upper Mantle Olivine and the Role of Titanium in the Water Weakening Mechanism. J. Geophys. Res. 2007, 112, B05211. [Google Scholar] [CrossRef] [Green Version]
- Balan, E.; Ingrin, J.; Delattre, S.; Kovács, I.; Blanchard, M. Theoretical infrared spectrum of OH-defects in forsterite. Eur. J. Miner. 2011, 23, 285–292. [Google Scholar] [CrossRef]
- Churakov, S.V.; Khisina, N.R.; Urusov, V.S.; Wirth, R. First-Principles Study of (MgH2SiO4)•n(Mg2SiO4) Hydrous Olivine Structures. I. Crystal Structure Modelling of Hydrous Olivine Hy-2a (MgH2SiO4) • 3(Mg2SiO4). Phys. Chem. Miner. 2003, 30, 1–11. [Google Scholar] [CrossRef]
- Blanchard, M.; Ingrin, J.; Balan, E.; Kovács, I.; Withers, A.C. Effect of Iron and Trivalent Cations on OH Defects in Olivine. Am. Mineral. 2017, 102, 302–311. [Google Scholar] [CrossRef]
- McGetchin, T.R.; Silver, L.T.; Chodos, A.A. Titanoclinohumite: A Possible Mineralogical Site for Water in the Upper Mantle. J. Geophys. Res. 1970, 75, 255–259. [Google Scholar] [CrossRef] [Green Version]
- Kohlstedt, D.L.; Mackwell, S.J. Diffusion of Hydrogen and Intrinsic Point Defects in Olivine. Z. Für Phys. Chem. 1998, 207, 147–162. [Google Scholar] [CrossRef]
- Schock, R.N.; Duba, A.G. Points Defects and the Mechanisms of Electrical Conduction in Olivine. Point Defects Miner. 2013, 31, 88–96. [Google Scholar] [CrossRef]
- Defects in Solids | Wiley. Available online: https://www.wiley.com/en-us/Defects+in+Solids-p-9780470380734 (accessed on 28 March 2023).
- Kramer, M.J.; Mendelev, M.I.; Napolitano, R.E. In Situ Observation of Antisite Defect Formation during Crystal Growth. Phys. Rev. Lett. 2010, 105, 245501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehr, K.T.; Hochleitner, R.; Schmidbauer, E.; Schneider, J. Mineralogy, Mössbauer Spectra and Electrical Conductivity of Triphylite Li(Fe2+, Mn2+) PO4. Phys. Chem. Miner. 2007, 34, 485–494. [Google Scholar] [CrossRef]
- Hatert, F.; Ottolini, L.; Wouters, J.; Fontan, F. A Structural Study of the Lithiophilite-Sicklerite Series. Can. Mineral. 2012, 50, 843–854. [Google Scholar] [CrossRef]
- Drozhzhin, O.A.; Tereshchenko, I.V.; Emerich, H.; Antipov, E.V.; Abakumov, A.M.; Chernyshov, D. An Electrochemical Cell with Sapphire Windows for Operando Synchrotron X-Ray Powder Diffraction and Spectroscopy Studies of High-Power and High-Voltage Electrodes for Metal-Ion Batteries. J. Synchrotron. Radiat. 2018, 25, 468–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.S.; Driscoll, D.J.; Fisher, C.A.J.; Slater, P.R. Atomic-Scale Investigation of Defects, Dopants, and Lithium Transport in the LiFePO4 Olivine-Type Battery Material. Chem. Mater. 2005, 17, 5085–5092. [Google Scholar] [CrossRef]
- Fisher, C.A.J.; Prieto, V.M.H.; Islam, M.S. Lithium Battery Materials LiMPO4 (M = Mn, Fe, Co, and Ni): Insights into Defect Association, Transport Mechanisms, and Doping Behavior. Available online: https://pubs.acs.org/doi/full/10.1021/cm801262x (accessed on 30 May 2023).
- Malik, R.; Burch, D.; Bazant, M.; Ceder, G. Particle Size Dependence of the Ionic Diffusivity. Available online: https://pubs.acs.org/doi/full/10.1021/nl1023595 (accessed on 30 May 2023).
- Tripathi, R.; Wood, S.M.; Islam, M.S.; Nazar, L.F. Na-Ion Mobility in Layered Na2FePO4F and Olivine Na[Fe, Mn]PO4. Energy Environ. Sci. 2013, 6, 2257–2264. [Google Scholar] [CrossRef]
- Galceran, M.; Roddatis, V.; Zúñiga, F.J.; Pérez-Mato, J.M.; Acebedo, B.; Arenal, R.; Peral, I.; Rojo, T.; Casas-Cabanas, M. Na–Vacancy and Charge Ordering in Na≈2/3FePO4. Chem. Mater. 2014, 26, 3289–3294. [Google Scholar] [CrossRef]
- Casas-Cabanas, M.; Roddatis, V.V.; Saurel, D.; Kubiak, P.; Carretero-González, J.; Palomares, V.; Serras, P.; Rojo, T. Crystal Chemistry of Na Insertion/Deinsertion in FePO4–NaFePO4. J. Mater. Chem. 2012, 22, 17421–17423. [Google Scholar] [CrossRef]
- Lee, J.; Zhou, W.; Idrobo, J.C.; Pennycook, S.J.; Pantelides, S.T. Vacancy-Driven Anisotropic Defect Distribution in the Battery-Cathode Material LiFePO4. Phys. Rev. Lett. 2011, 107, 085507. [Google Scholar] [CrossRef] [Green Version]
- Kuss, C.; Liang, G.; Schougaard, S.B. Atomistic Modeling of Site Exchange Defects in Lithium Iron Phosphate and Iron Phosphate. J. Mater. Chem. 2012, 22, 24889–24893. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Sun, X. Olivine LiFePO4: The Remaining Challenges for Future Energy Storage. Energy Environ. Sci. 2015, 8, 1110–1138. [Google Scholar] [CrossRef]
- Hydrothermal Synthesis of Lithium Iron Phosphate Cathodes. Electrochem. Commun. 2001, 3, 505–508. [CrossRef]
- Cho, M.-Y.; Kim, K.-B.; Lee, J.-W.; Kim, H.; Kim, H.; Kang, K.; Roh, K.C. Defect-Free Solvothermally Assisted Synthesis of Microspherical Mesoporous LiFePO4/C. RSC Adv. 2013, 3, 3421–3427. [Google Scholar] [CrossRef]
- Yang, S.; Song, Y.; Zavalij, P.Y.; Stanley Whittingham, M. Reactivity, Stability and Electrochemical Behavior of Lithium Iron Phosphates. Electrochem. Commun. 2002, 4, 239–244. [Google Scholar] [CrossRef]
- Chen, J.; Vacchio, M.J.; Wang, S.; Chernova, N.; Zavalij, P.Y.; Whittingham, M.S. The Hydrothermal Synthesis and Characterization of Olivines and Related Compounds for Electrochemical Applications. Solid State Ion. 2008, 178, 1676–1693. [Google Scholar] [CrossRef]
- Park, K.-Y.; Park, I.; Kim, H.; Lim, H.; Hong, J.; Kim, J.; Kang, K. Anti-Site Reordering in LiFePO4: Defect Annihilation on Charge Carrier Injection. Available online: https://pubs.acs.org/doi/full/10.1021/cm502432q (accessed on 30 May 2023).
- Chen, J.; Graetz, J. Study of Antisite Defects in Hydrothermally Prepared LiFePO4 by in Situ X-Ray Diffraction. Available online: https://pubs.acs.org/doi/full/10.1021/am200141a (accessed on 30 May 2023).
- Gibot, P.; Casas-Cabanas, M.; Laffont, L.; Levasseur, S.; Carlach, P.; Hamelet, S.; Tarascon, J.-M.; Masquelier, C. Room-Temperature Single-Phase Li Insertion/Extraction in Nanoscale LixFePO4. Nat. Mater 2008, 7, 741–747. [Google Scholar] [CrossRef]
- Yamada, A.; Koizumi, H.; Sonoyama, N.; Kanno, R. Phase Change in LixFePO4. Electrochem. Solid-State Lett. 2005, 8, A409. [Google Scholar] [CrossRef]
- Delacourt, C.; Poizot, P.; Levasseur, S.; Masquelier, C. Size Effects on Carbon-Free LiFePO4 Powders: The Key to Superior Energy Density. Electrochem. Solid-State Lett. 2006, 9, A352. [Google Scholar] [CrossRef]
- Zhao, Y.; Peng, L.; Liu, B.; Yu, G. Single-Crystalline LiFePO4 Nanosheets for High-Rate Li-Ion Batteries. Available online: https://pubs.acs.org/doi/full/10.1021/nl5008568 (accessed on 30 May 2023).
- Liang, Y.; Wen, K.; Mao, Y.; Liu, Z.; Zhu, G.; Yang, F.; He, W. Shape and Size Control of LiFePO4 for High-Performance Lithium-Ion Batteries. ChemElectroChem 2015, 2, 1227–1237. [Google Scholar] [CrossRef]
- Paolella, A.; Turner, S.; Bertoni, G.; Hovington, P.; Flacau, R.; Boyer, C.; Feng, Z.; Colombo, M.; Marras, S.; Prato, M.; et al. Accelerated Removal of Fe-Antisite Defects while Nanosizing Hydrothermal LiFePO4 with Ca2+. Available online: https://pubs.acs.org/doi/full/10.1021/acs.nanolett.6b00334 (accessed on 30 May 2023).
- Zou, Y.; Chang, G.; Chen, S.; Liu, T.; Xia, Y.; Chen, C.; Yang, D. Alginate/r-GO Assisted Synthesis of Ultrathin LiFePO4 Nanosheets with Oriented (0 1 0) Facet and Ultralow Antisite Defect. Chem. Eng. J. 2018, 351, 340–347. [Google Scholar] [CrossRef]
- Drozhzhin, O.A.; Sumanov, V.D.; Karakulina, O.M.; Abakumov, A.M.; Hadermann, J.; Baranov, A.N.; Stevenson, K.J.; Antipov, E.V. Switching between Solid Solution and Two-Phase Regimes in the Li1-XFe1-YMnyPO4 Cathode Materials during Lithium (de)Insertion: Combined PITT, in Situ XRPD and Electron Diffraction Tomography Study. Electrochim. Acta 2016, 191, 149–157. [Google Scholar] [CrossRef]
- Chung, S.-Y.; Choi, S.-Y.; Yamamoto, T.; Ikuhara, Y. Orientation-Dependent Arrangement of Antisite Defects in Lithium Iron(II) Phosphate Crystals. Angew. Chem. Int. Ed. 2009, 48, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.-Y.; Choi, S.-Y.; Yamamoto, T.; Ikuhara, Y. Atomic-Scale Visualization of Antisite Defects in LiFePO4. Phys. Rev. Lett. 2008, 100, 125502. [Google Scholar] [CrossRef]
- Chapman, C.A. Large Magnesia-Rich Triphylite Crystals in Pegmatite. Am. Mineral. 1943, 28, 90–98. [Google Scholar]
- Tealdi, C.; Spreafico, C.; Mustarelli, P. Lithium Diffusion in Li1−xFePO4: The Effect of Cationic Disorder. J. Mater. Chem. 2012, 22, 24870–24876. [Google Scholar] [CrossRef]
- Boulfelfel, S.E.; Seifert, G.; Leoni, S. Atomistic Investigation of Li+ Diffusion Pathways in the Olivine LiFePO4 Cathode Material. J. Mater. Chem. 2011, 21, 16365–16372. [Google Scholar] [CrossRef]
- Chung, S.-Y.; Choi, S.-Y.; Lee, S.; Ikuhara, Y. Distinct Configurations of Antisite Defects in Ordered Metal Phosphates: Comparison between LiMnPO4 and LiFePO4. Phys. Rev. Lett. 2012, 108, 195501. [Google Scholar] [CrossRef] [Green Version]
- Frencis, C.A. The Forsterite-Tephroite Series: I Crystal Structure Refinements. Available online: https://www.semanticscholar.org/paper/The-forsterite-tephroite-series-%3A-I-.-Crystal-FReNcIs/b6c28c0f2f8accb9386cdf33ff498a28584362a0 (accessed on 30 May 2023).
- Park, K.-Y.; Park, I.; Kim, H.; Yoon, G.; Gwon, H.; Cho, Y.; SooYun, Y.; Kim, J.-J.; Lee, S.; Ahn, D.; et al. Lithium-Excess Olivine Electrode for Lithium Rechargeable Batteries. Energy Environ. Sci. 2016, 9, 2902–2915. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Gu, Y.; Teng, G.; Liu, Y.; Zheng, J.; Pan, F. Ab Initio Identification of the Li-Rich Phase in LiFePO4. Phys. Chem. Chem. Phys. 2018, 20, 17497–17503. [Google Scholar] [CrossRef]
- Janssen, Y.; Santhanagopalan, D.; Qian, D.; Chi, M.; Wang, X.; Hoffmann, C.; Meng, Y.S.; Khalifah, P.G. Reciprocal Salt Flux Growth of LiFePO4 Single Crystals with Controlled Defect Concentrations. Chem. Mater. 2013, 25, 4574–4584. [Google Scholar] [CrossRef] [Green Version]
- Albertia, A. The Crystal Structure of Ferrisicklerite, Li<1(Fe3+,Mn2+)PO4. Acta. Cryst. B 1976, 32, 2761–2764. [Google Scholar] [CrossRef]
- Amisse, R.; Sougrati, M.T.; Stievano, L.; Davoisne, C.; Dražič, G.; Budič, B.; Dominko, R.; Masquelier, C. Singular Structural and Electrochemical Properties in Highly Defective LiFePO4 Powders. Chem. Mater. 2015, 27, 4261–4273. [Google Scholar] [CrossRef]
- Ong, S.P.; Wang, L.; Kang, B.; Ceder, G. Li−Fe−P−O2 Phase Diagram from First Principles Calculations. Chem. Mater. 2008, 20, 1798–1807. [Google Scholar] [CrossRef]
- Asmare, A. Brodie Whitehead Thermodynamic Principles of Dittmarite Precipitation. JESE-B 2018, 7, 331–336. [Google Scholar] [CrossRef]
- Koleva, V.; Stoyanova, R.; Zhecheva, E.; Nihtianova, D. Dittmarite Precursors for Structure and Morphology Directed Synthesis of Lithium Manganese Phospho-Olivine Nanostructures. CrystEngComm 2014, 16, 7515–7524. [Google Scholar] [CrossRef]
- Eventoff, W.; Martin, R.; Peacor, D.R. The Crystal Structure of Heterosite1. Am. Mineral. 1972, 57, 45–51. [Google Scholar]
- Pramanik, A.; Bradford, A.J.; Lee, S.L.; Lightfoot, P.; Armstrong, A.R. Na2Fe(C2O4)(HPO4): A Promising New Oxalate-Phosphate Based Mixed Polyanionic Cathode for Li/Na Ion Batteries. J. Phys. Mater. 2021, 4, 024004. [Google Scholar] [CrossRef]







| Atomic Coordinates * | |||||
|---|---|---|---|---|---|
| Structure Type | Olivine (M2XO4) [20] | Triphylite (LiMPO4) [7] | Maricite (NaMPO4) [21] | ||
| Wyckoff Position | M1 | 4a | 0, 0, 0 | 0, 0, 0 | 0, 0, 0 |
| M2 | 4c | 0.28, ¼, 0.98 | 0.28, ¼, 0.97 | 0.34, ¼, 0.97 | |
| T | 4c | 0.10, ¼, 0.40 | 0.09, ¼, 0.41 | 0.32, ¼, 0.04 | |
| O1 | 4c | 0.08, ¼, 0.75 | 0.09, ¼, 0.73 | 0.12, ¼, 0.75 | |
| O2 | 4c | 0.42, ¼, 0.25 | 0.45, ¼, 0.21 | 0.34, ¼, 0.45 | |
| O3 | 8d | 0.17, 0.01, 0.25 | 0.16, 0.05, 0.28 | 0.12, 0.06, 0.32 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nazarov, E.E.; Aksyonov, D.A.; Antipov, E.V.; Fedotov, S.S. A Comprehensive Review of Defects Genealogy in Olivines: From the Mineral World to Modern Electrode Materials. Energies 2023, 16, 5083. https://doi.org/10.3390/en16135083
Nazarov EE, Aksyonov DA, Antipov EV, Fedotov SS. A Comprehensive Review of Defects Genealogy in Olivines: From the Mineral World to Modern Electrode Materials. Energies. 2023; 16(13):5083. https://doi.org/10.3390/en16135083
Chicago/Turabian StyleNazarov, Eugene E., Dmitry A. Aksyonov, Evgeny V. Antipov, and Stanislav S. Fedotov. 2023. "A Comprehensive Review of Defects Genealogy in Olivines: From the Mineral World to Modern Electrode Materials" Energies 16, no. 13: 5083. https://doi.org/10.3390/en16135083