
A First-Principles Study on Titanium-Decorated Adsorbent for Hydrogen Storage

 ,
,
Abstract
:1. Introduction
2. Computational Methods
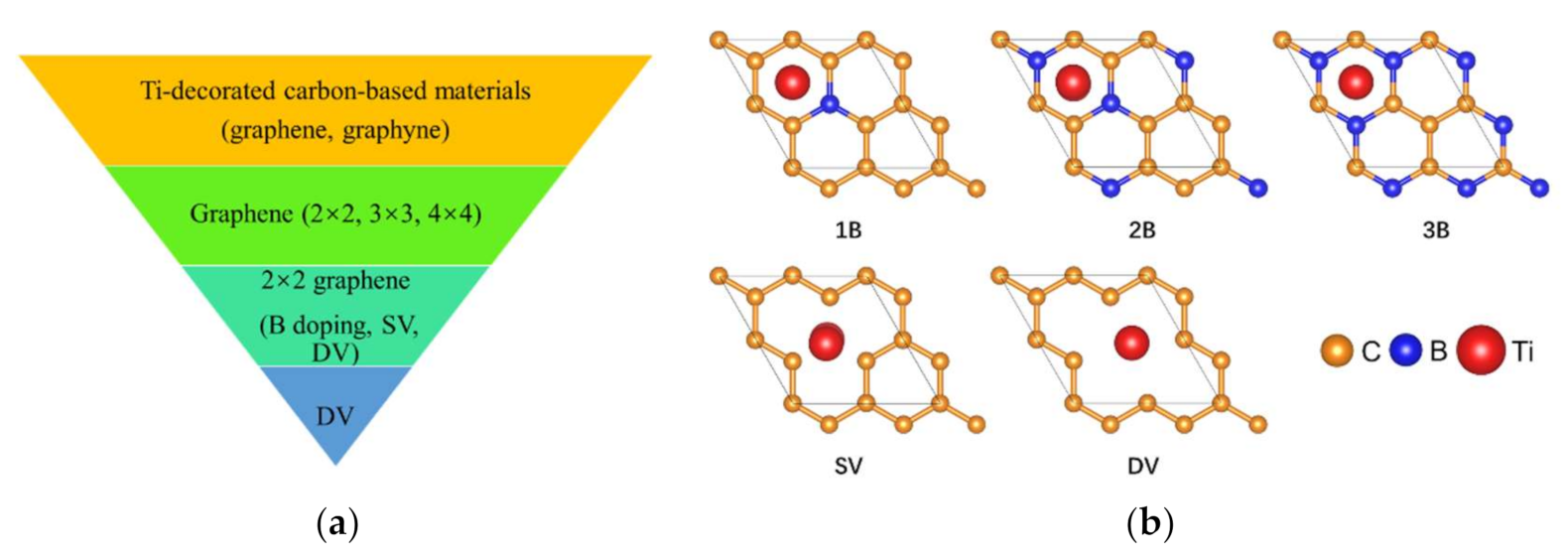
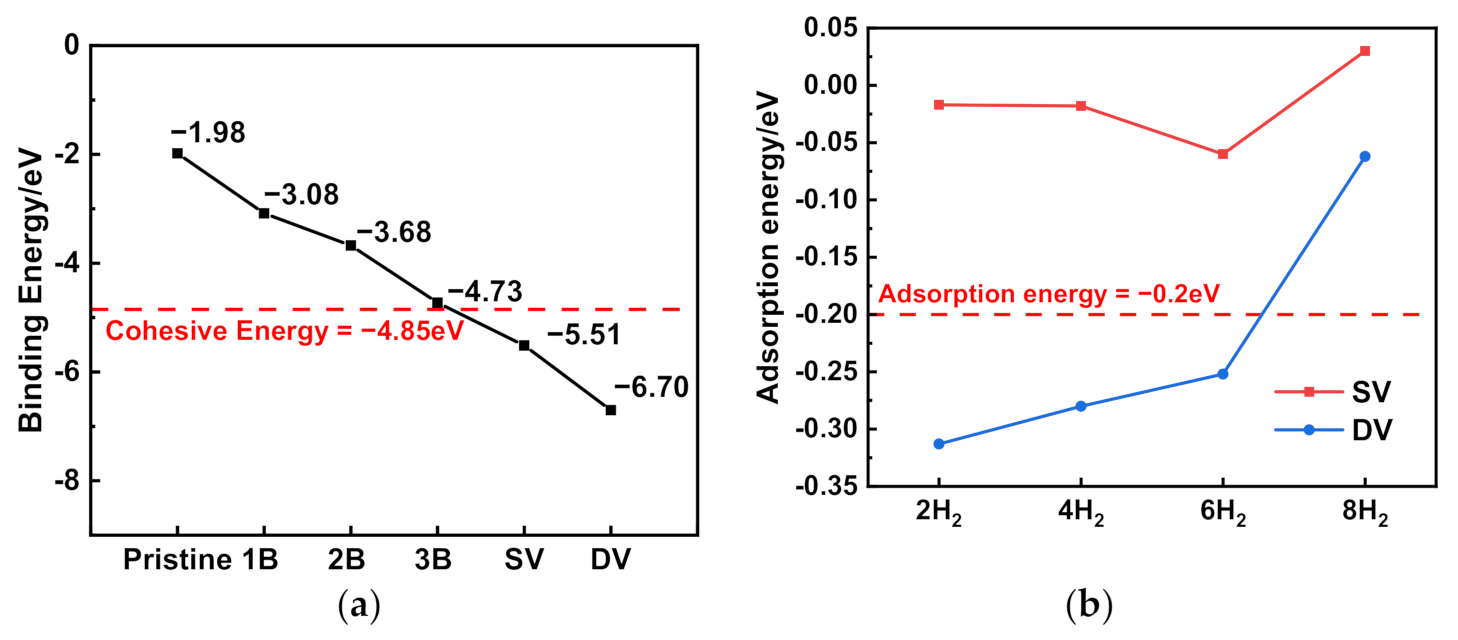
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schlapbach, L.; Züttel, A. Hydrogen-storage materials for mobile application. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef]
- Züttel, A. Materials for hydrogen storage. Mater. Today 2003, 6, 24–33. [Google Scholar] [CrossRef]
- Ding, Z.; Yang, W.; Huo, K.; Shaw, L. Thermodynamics and Kinetics Tuning of LiBH4 for Hydrogen Storage. Prog. Chem. 2020, 64. [Google Scholar] [CrossRef]
- Andersson, J.; Grönkvist, S. Large-scale storage of hydrogen. Int. J. Hydrogen Energy 2019, 44, 11901–11919. [Google Scholar] [CrossRef]
- U.S. Department of Energy. 2017. Available online: www.energy.gov/eere/fuelcells/hydrogen-storage (accessed on 8 June 2021).
- Jena, P. Materials for hydrogen storage: Past, present, and future. J. Phys. Chem. Lett. 2011, 2, 206–211. [Google Scholar] [CrossRef]
- Dillon, A.C.; Jones, K.M.; Bekkedahl, T.A.; Kiang, C.H.; Bethune, D.S.; Heben, M.J. Storage of hydrogen in single-walled carbon nanotubes. Nature 1997, 386, 377–379. [Google Scholar] [CrossRef]
- Yoon, M.; Yang, S.; Hicke, C.; Wang, E.; Geohegan, D.; Zhang, Z. Calcium as the superior coating metal in functionalization of carbon fullerenes for high-capacity hydrogen storage. Phys. Rev. Lett. 2008, 100, 206806. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Ihm, J.; Cohen, M.L.; Louie, S.G. Calcium-decorated carbon nanotubes for high-capacity hydrogen storage: First-principles calculations. Phys. Rev. B 2009, 80, 115412. [Google Scholar] [CrossRef]
- López-Corral, I.; Germán, E.; Juan, A.; Volpe, M.A.; Brizuela, G.P. DFT study of hydrogen adsorption on palladium decorated graphene. J. Phys. Chem. C 2011, 115, 4315–4323. [Google Scholar] [CrossRef]
- Xiao, H.; Li, S.H.; Cao, J.X. First-principles study of Pd-decorated carbon nanotube for hydrogen storage. Chem. Phys. Lett. 2009, 483, 111–114. [Google Scholar] [CrossRef]
- Ao, Z.M.; Jiang, Q.; Zhang, R.Q.; Tan, T.T.; Li, S. Al doped graphene: A promising material for hydrogen storage at room temperature. J. Appl. Phys. 2009, 105, 074307. [Google Scholar] [CrossRef] [Green Version]
- Ataca, C.; Akturk, E.; Ciraci, S.; Ustunel, H. High-capacity hydrogen storage by metallized graphene. Appl. Phys. Lett. 2008, 93, 043123. [Google Scholar] [CrossRef]
- Ataca, C.; Aktürk, E.; Ciraci, S. Hydrogen storage of calcium atoms adsorbed on graphene: First-principles plane wave calculations. Phys. Rev. B 2009, 79, 041406. [Google Scholar] [CrossRef]
- Tozzini, V.; Pellegrini, V. Prospects for hydrogen storage in graphene. Phys. Chem. Chem. Phys. 2013, 15, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Ng, S.-P.; Ding, N.; Wu, C.-M.L. Strain-induced switch for hydrogen storage in cobalt-decorated nitrogen-doped graphene. Appl. Surf. Sci. 2019, 473, 174–181. [Google Scholar] [CrossRef]
- Chu, S.; Hu, L.; Hu, X.; Yang, M.; Deng, J. Titanium-embedded graphene as high-capacity hydrogen-storage media. Int. J. Hydrog. Energy 2011, 36, 12324–12328. [Google Scholar] [CrossRef]
- Jain, V.; Kandasubramanian, B. Functionalized graphene materials for hydrogen storage. J. Mater. Sci. 2019, 55, 1865–1903. [Google Scholar] [CrossRef]
- Ao, Z.M.; Dou, S.X.; Xu, Z.M.; Jiang, Q.G.; Wang, G.X. Hydrogen storage in porous graphene with Al decoration. Int. J. Hydrog. Energy 2014, 39, 16244–16251. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, T.; Hou, X.; Zhang, W.; Tang, S.; Sun, H.; Zhang, J. Li-decorated porous graphene as a high-performance hydrogen storage material: A first-principles study. Int. J. Hydrog. Energy 2017, 42, 10099–10108. [Google Scholar] [CrossRef]
- Wu, M.; Gao, Y.; Zhang, Z.; Zeng, X.C. Edge-decorated graphene nanoribbons by scandium as hydrogen storage media. Nanoscale 2012, 4, 915–920. [Google Scholar] [CrossRef]
- Lebon, A.; Carrete, J.; Gallego, L.J.; Vega, A. Ti-decorated zigzag graphene nanoribbons for hydrogen storage. A van der Waals-corrected density-functional study. Int. J. Hydrog. Energy 2015, 40, 4960–4968. [Google Scholar] [CrossRef]
- Chan, S.-P.; Chen, G.; Gong, X.G.; Liu, Z.-F. Chemisorption of hydrogen molecules on carbon nanotubes under high pressure. Phys. Rev. Lett. 2001, 87, 205502. [Google Scholar] [CrossRef]
- Reunchan, P.; Jhi, S.-H. Metal-dispersed porous graphene for hydrogen storage. Appl. Phys. Lett. 2011, 98, 093103. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Wang, D.; Gong, J.; Zhang, C.; Zhang, L.; Zhang, M.; Wu, X.; Kang, L. First-principles study of V-decorated porous graphene for hydrogen storage. Chem. Phys. Lett. 2019, 726, 57–61. [Google Scholar] [CrossRef]
- Chakraborty, B.; Modak, P.; Banerjee, S. Hydrogen storage in yttrium-decorated single walled carbon nanotube. J. Phys. Chem. C 2012, 116, 22502–22508. [Google Scholar] [CrossRef]
- Yildirim, T.; Ciraci, S. Titanium-decorated carbon nanotubes as a potential high-capacity hydrogen storage medium. Phys. Rev. Lett. 2005, 94, 175501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.; Rao, D.; Meng, Z.; Zhang, X.; Xu, G.; Liu, Y.; Kan, E.; Xiao, C.; Deng, K. Boron-substituted graphyne as a versatile material with high storage capacities of Li and H2: A multiscale theoretical study. Phys. Chem. Chem. Phys. 2013, 15, 16120–16126. [Google Scholar] [CrossRef]
- Hussain, T.; Pathak, B.; Ramzan, M.; Maark, T.A.; Ahuja, R. Calcium doped graphane as a hydrogen storage material. Appl. Phys. Lett. 2012, 100, 183902. [Google Scholar] [CrossRef]
- Ao, Z.M.; Peeters, F.M. High-capacity hydrogen storage in Al-adsorbed graphene. Phys. Rev. B 2010, 81, 205406. [Google Scholar] [CrossRef]
- Zheng, N.; Yang, S.; Xu, H.; Lan, Z.; Wang, Z.; Gu, H. A DFT study of the enhanced hydrogen storage performance of the Li-decorated graphene nanoribbons. Vacuum 2020, 171, 109011. [Google Scholar] [CrossRef]
- Ramos-Castillo, C.M.; Reveles, J.U.; Cifuentes-Quintal, M.E.; Zope, R.R.; de Coss, R. Ti4- and Ni4-Doped Defective Graphene Nanoplatelets as Efficient Materials for Hydrogen Storage. J. Phys. Chem. C 2016, 120, 5001–5009. [Google Scholar] [CrossRef]
- Liu, Y.; Ren, L.; He, Y.; Cheng, H.P. Titanium-decorated graphene for high-capacity hydrogen storage studied by density functional simulations. J. Phys. Condens. Matter 2010, 22, 445301. [Google Scholar] [CrossRef]
- Park, H.L.; Yoo, D.S.; Yi, S.C.; Chung, Y.C. Theoretical investigation of Ti-adsorbed graphene for hydrogen storage using the ab-initio method. J. Nanosci. Nanotechnol. 2011, 11, 6131–6135. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Xu, S.; Ma, K.; Wu, C.; Gates, I.D.; Ding, X.; Meng, W.; Gao, Z. Geometric structures, electronic characteristics, stabilities, catalytic activities, and descriptors of graphene-based single-atom catalysts. Nano Mater. Sci. 2020, 2, 120–131. [Google Scholar] [CrossRef]
- Yoon, M.; Yang, S.; Wang, E.; Zhang, Z. Charged fullerenes as high-capacity hydrogen storage media. Nano Lett. 2007, 7, 2578–2583. [Google Scholar] [CrossRef]
- Krasheninnikov, A.V.; Lehtinen, P.O.; Foster, A.S.; Pyykko, P.; Nieminen, R.M. Embedding transition-metal atoms in graphene: Structure, bonding, and magnetism. Phys. Rev. Lett. 2009, 102, 126807. [Google Scholar] [CrossRef] [Green Version]
- Kubas, G.J. Metal–dihydrogen and σ-bond coordination: The consummate extension of the Dewar–Chatt–Duncanson model for metal–olefin π bonding. J. Organomet. Chem. 2001, 635, 37–68. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calculation Details | Parameter |
|---|---|
| spin polarization | ISPIN = 2 |
| energy cutoff/eV | ENCUT = 450.0 |
| convergence condition (energy)/eV | EDIFF = 1 × 10−5 |
| convergence condition (force)/eV/Å | EDIFFG = −0.05 |
| dipole correction | LDIPOL = TRUE. IDIPOL = 3 |
| degrees-of-freedom | ISIF = 2 |
| van der Waals interactions | IVDW = 12 |
| k-point | 6 × 6 × 1 |
| Number of H2 | Adsorption Energy | Bond Length of H-H (Up) | Bond Length of H-H (Down) |
|---|---|---|---|
| 2 H2 | −0.3134 | 0.804 | 0.814 |
| 4 H2 | −0.2802 | 0.804 0.800 | 0.804 0.814 |
| 6 H2 | −0.2519 | 0.804 0.800 0.786 | 0.804 0.814 0.789 |
| 8 H2 | −0.0626 | 0.788 0.783 0.784 0.785 | 0.789 0.784 0.782 0.784 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, K.; Lv, E.; Zheng, D.; Cui, W.; Dong, S.; Yang, W.; Gao, Z.; Zhou, Y. A First-Principles Study on Titanium-Decorated Adsorbent for Hydrogen Storage. Energies 2021, 14, 6845. https://doi.org/10.3390/en14206845
Ma K, Lv E, Zheng D, Cui W, Dong S, Yang W, Gao Z, Zhou Y. A First-Principles Study on Titanium-Decorated Adsorbent for Hydrogen Storage. Energies. 2021; 14(20):6845. https://doi.org/10.3390/en14206845
Chicago/Turabian StyleMa, Kai, Erfei Lv, Di Zheng, Weichun Cui, Shuai Dong, Weijie Yang, Zhengyang Gao, and Yu Zhou. 2021. "A First-Principles Study on Titanium-Decorated Adsorbent for Hydrogen Storage" Energies 14, no. 20: 6845. https://doi.org/10.3390/en14206845