1. Introduction
This paper considers the task of estimating the energy and power density of LIB designs across a range of characteristics and parameters and focuses on the problem of obtaining these results without incurring the considerable computational costs of detailed physical simulations in a recurrent manner. While state-of-the-art contributions such as [
1] exploit Deep Learning neural networks trained on simulated data as surrogate models for this objective, to the best of our knowledge, no literature contribution yet has evaluated the performance of so-called ensemble models for this task. We address this gap in the literature by comparing state-of-the-art models with a number of ensemble surrogates.
Even though Lithium-Ion Batteries (LIB) have progressively been improved since their market introduction in 1991 by Sony, their massive deployment requires them to be further optimized in terms of performance, durability, and safety. From the physical point of view, to obtain these results, in the battery design phase, it is important to reduce limitations to ion transport, thus avoiding undesirable cell polarization effects. Consequently, an extensive literature describes the effect each design parameter has on transport mechanisms, and therefore on cell performance. On the one hand, the effects of several parameters (including electrode thickness, particle size, tortuosity and discharge rate) have been investigated by experimental methods. On the other hand, cell design has also been explored through modeling approaches, demonstrating that model-based design can be used to reduce the number of experiments, while accurately describing battery performance and providing guidance for battery characterization.
In general, the battery models presented in the literature mainly fall into two categories: physics-based electrochemical models and empirical ones. In addition, the recent development of physics-based equivalent circuit models aims to combine the high accuracy of physical models with the reduced computational cost of empirical ones.
Physics-based electrochemical models [
2,
3,
4] use partial differential equations to describe the phenomena taking place within the battery. They can be used to forecast its electrochemical state, and to provide accurate information about variables such as lithium concentrations and over-potentials, which can be used to understand the phenomena that are limiting performance or durability. For example, the widely used Pseudo-two-Dimensional (P2D) electrochemical model developed by Doyle and Newman [
5,
6] is based on porous electrode theory, concentrated solution theory, and kinetics equations. Further electrochemical modeling approaches rely on Single Particle (SP) simplifications, in which the properties of the electrolyte are normally ignored [
7,
8,
9]. Further recent approaches such as SPMe [
10] consider electrolyte dynamics in SP models. Other possibilities include Simplified Models building on polynomial profiles [
11,
12], Galerkin approximations [
13], transfer function modeling [
14], and the like. Concerning limitations, a drawback of detailed electrochemical models is the significant computational cost of simulations [
15]. To overcome this limitation, various reduced-order models have been developed, which are either distributed-parameter models [
10,
13] or lumped-parameters models [
14,
16]. Distributed-parameter models are normally expressed in the form of ordinary differential-algebraic equations (DAEs), derived, for example, using the Galerkin projection methods or Proper Orthogonal Decomposition, trying to preserve the physical meaning of all model parameters. Lumped-parameter models, which mimic the output voltage of the battery using electrical components such as resistors and capacitors, are described below.
Empirical models are typically either based on equivalent circuits [
16,
17] or on data-driven approaches [
18,
19]. battery control algorithm rather than in cell design optimization applications. They are composed of an open-circuit voltage source connected to a set of electric elements, such as resistors and capacitors, to model the electrical behavior of a battery. While these models are intuitive and relatively simple to use in control system design and implementation, they do not provide insights on the internal behavior of the battery. In this regard, new approaches have recently been explored, such as the developed distributed-parameter ECMs [
20,
21], in which the models are normally expressed in the form of DAEs, which can be solved rapidly using the proposed method with high accuracy. This represents an improvement on existing physics-based Li-ion battery models, especially in real-time, dynamic environments. Physics-based equivalent circuit models combine the benefits of high accuracy physical models with the lower computational cost of empirical ones, for instance, by combining a concise transmission line structure with partial differential equations for the mass transport processes that describe the concentration distributions and that are solved with the finite difference method, avoiding simplifications or approximations, thus guaranteeing the accuracy of the results [
20]. Online estimation and prediction of the Remaining Useful Life also often use data-driven empirical methods which, however, have not been commonly exploited for cell design purposes.
So-called ‘surrogate’ models are obtained by Machine Learning methods from data generated by simulations. Once the training phase is completed, these models can provide estimations of battery performance indicators with a lower computational cost and with an accuracy similar to that of physical simulators. Probabilistic surrogate models are often used for optimization: by running the simulations at a set of points (experimental design), one obtains fast surrogates for otherwise expensive objective functions [
22]. In methods such as Upper and Lower Confidence Bound [
23], Expected Improvement [
24], DYCORS [
25], and SOP [
26], the optimization jointly performs both exploration and exploitation, looking for an optimum, while, at the same time, sampling by simulation the most uncertain parameter regions. In this sense, surrogate optimization schemes can efficiently leverage the ability of probabilistic surrogate model classes, from Gaussian Processes to Sequential Radial Basis Functions, to jointly generate estimates for both the local value and the local uncertainty of an objective function of interest. Relevant efforts for LIB aging modeling include those based on Gaussian processes by Liu et al. [
27,
28,
29].
Contributing to the domain of such surrogates, Wu et al. [
1] propose to address cell design characterization by combining Machine Learning classifiers and regressors based on deep feed-forward neural network models. While the first neural network is a classifier that predicts whether a set of input design variables would result in a physically realizable cell, the second neural network estimates the specific energy and specific power of the design. Both neural networks are trained and validated using data from finite-element, thermo-electrochemical simulations.
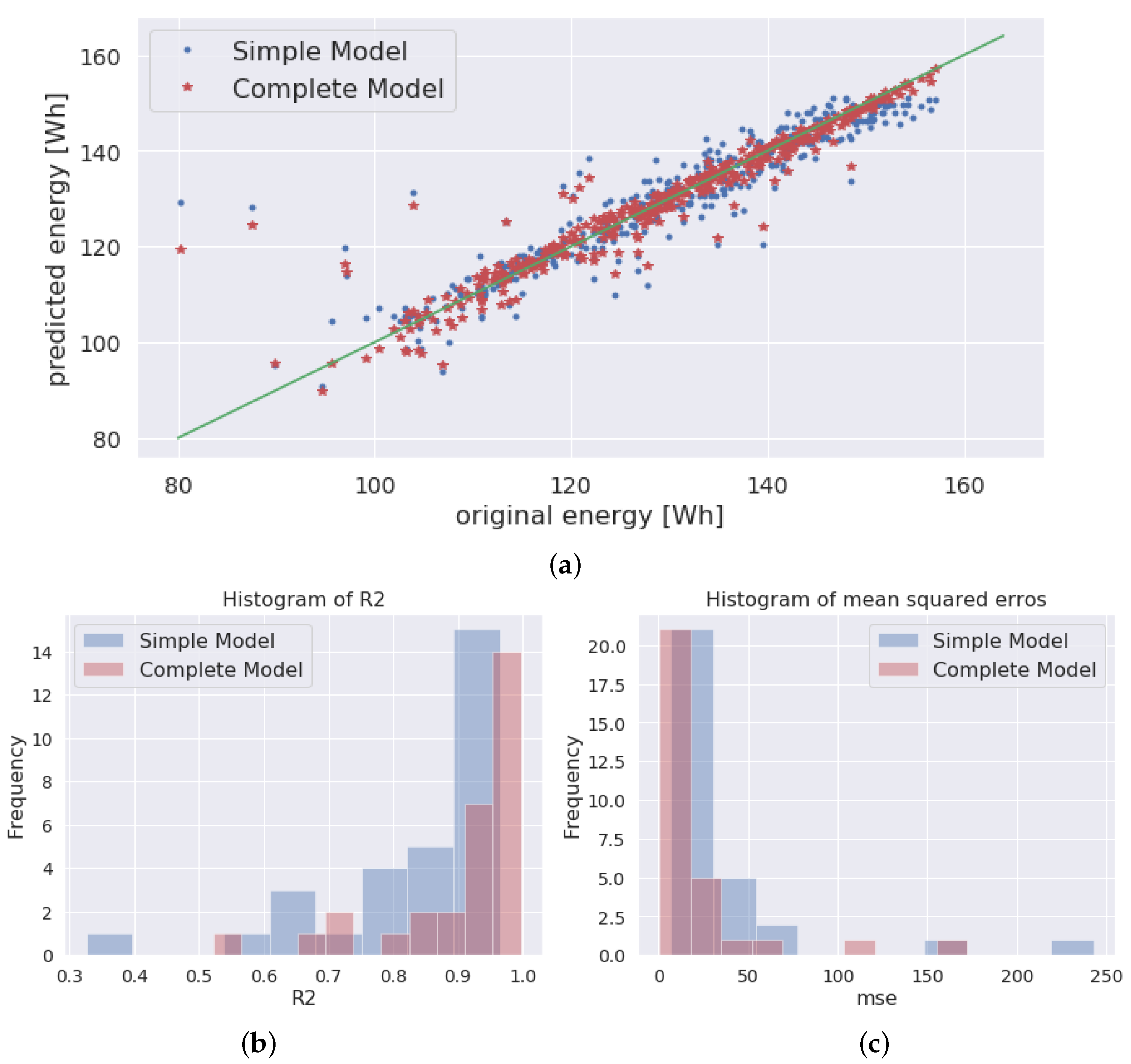
Although ensemble models are well represented in the literature, to the best of our knowledge, no contribution yet has evaluated their performance as surrogates of physical LIB simulators. This paper addresses this gap by comparing a state-of-the-art LIB surrogate model based on deep feed-forward networks with a set of ensemble models integrating deep classifiers and regressors. In this sense, we demonstrate the accuracy of composite surrogate models for the estimation of the density of power and energy of a given LIB parameter set. The input and output parameters taken into consideration for the electrochemical simulations are listed in
Table 1. Their values are measured by proprietary electrochemical and physicochemical protocols for a set of proprietary electrodes in CIDETEC, and vary between the minimum and maximum values shown in
Table 1.
In terms of application, the present paper does not directly focus on optimization. Instead, as also previously done in [
1], it considers surrogates that can be used to estimate the performance of battery designs across a wide range of material characteristics and parameters. This applicative objective reduces the importance of using surrogate models that can output estimated values and their uncertainties, for instance by exploiting techniques such as Monte Carlo Dropout [
30] and Variational Inference [
31]. Consequently, in the rest of our treatment, we limit ourselves to non-probabilistic surrogates.
The methodology we introduce and detail in the sections below is based on the general framework put forward by [
32,
33], integrating and extending the Pseudo-2D surrogate proposed by [
1] to evaluate the performance of composite surrogate models integrating both classification and regression. Through classification, a Machine Learning model evaluates the convergence of the simulator, while, through regression, the algorithms predict the output parameters of the Ragone plot [
34] (energy and power, as per
Figure 1). We extend the state of the art in [
1] by showing the advantage of adopting respectively ensemble methods for convergence classification and structured regression for the estimation of the Ragone parameters, rather than simpler feed-forward networks for both tasks. Furthermore, from the methodological point of view, we apply a quantitative performance evaluation procedure that is based on K-fold validation [
35] rather than on simple train/test/validation splitting as in [
1]. The accuracy of some of the ensemble surrogate models we introduce compares favorably to that of the state-of-the-art method introduced in [
1]. Furthermore, the approach in the reference is extended by using a K-fold cross-validation method to evaluate if the model can generalize the quality of its results.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}