Kinetics and Reactor Design Aspects of Selective Methanation of CO over a Ru/γ-Al2O3 Catalyst in CO2/H2 Rich Gases †

Abstract
:1. Introduction
2. Experimental
2.1. Catalyst Preparation and Characterization
2.2. Experimental Set-up
2.3. Kinetic Measurements
3. Results
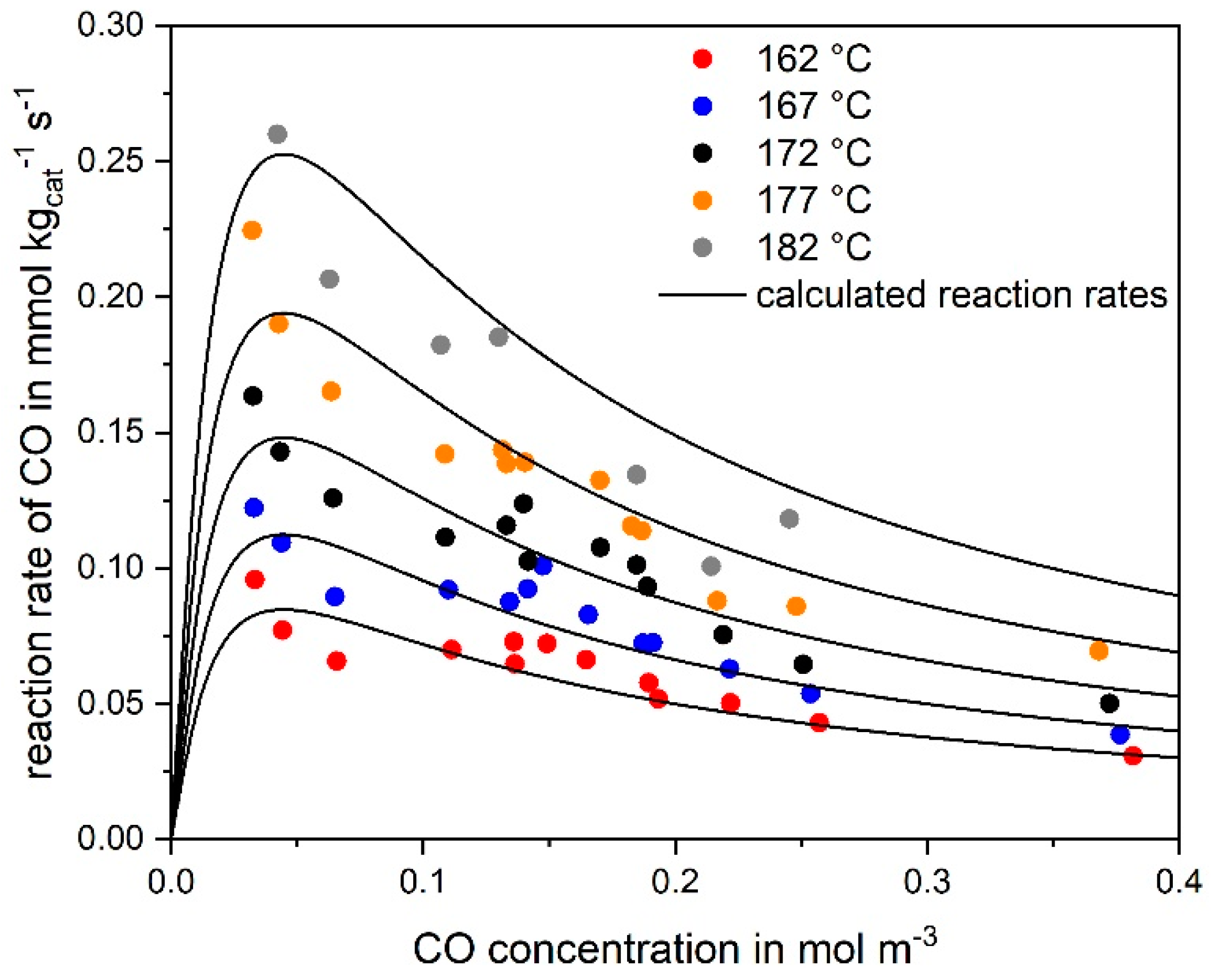
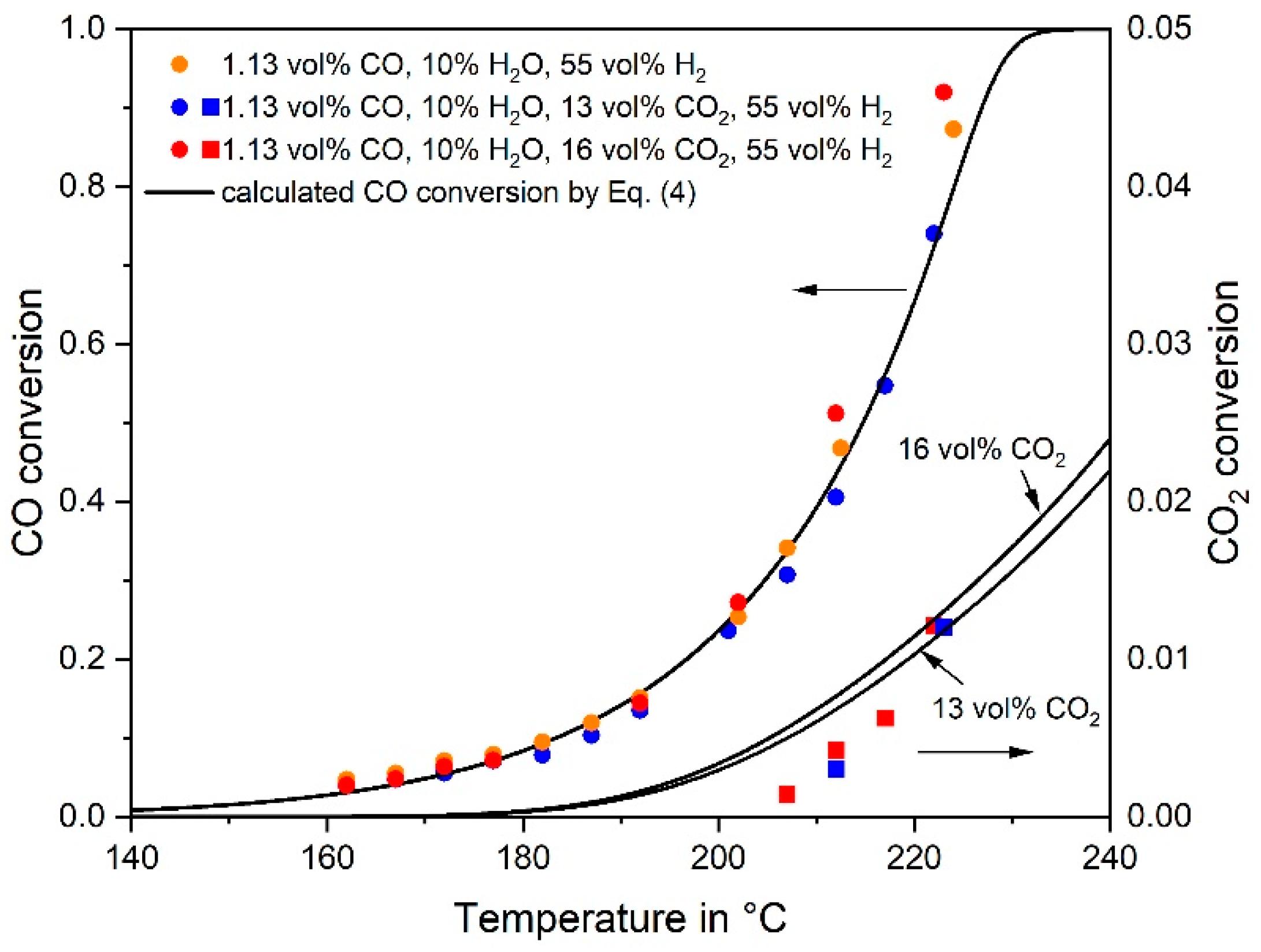
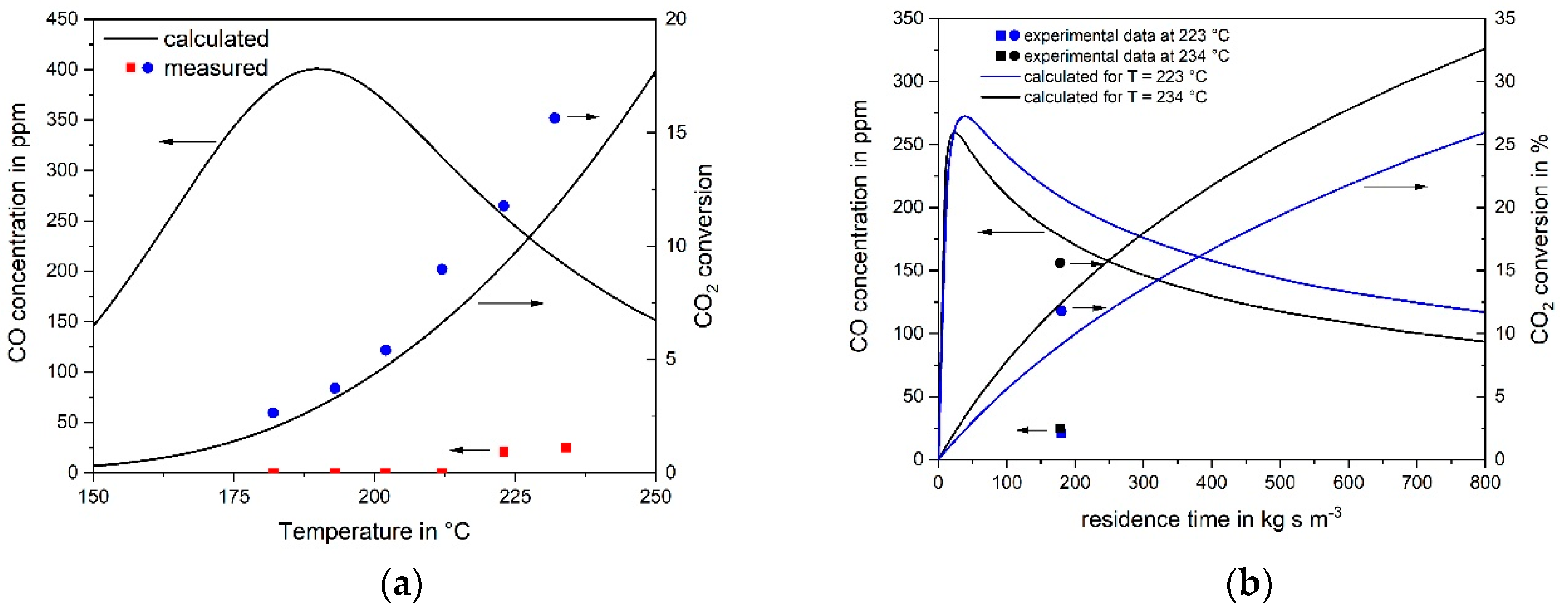
3.1. CO Methanation Kinetics
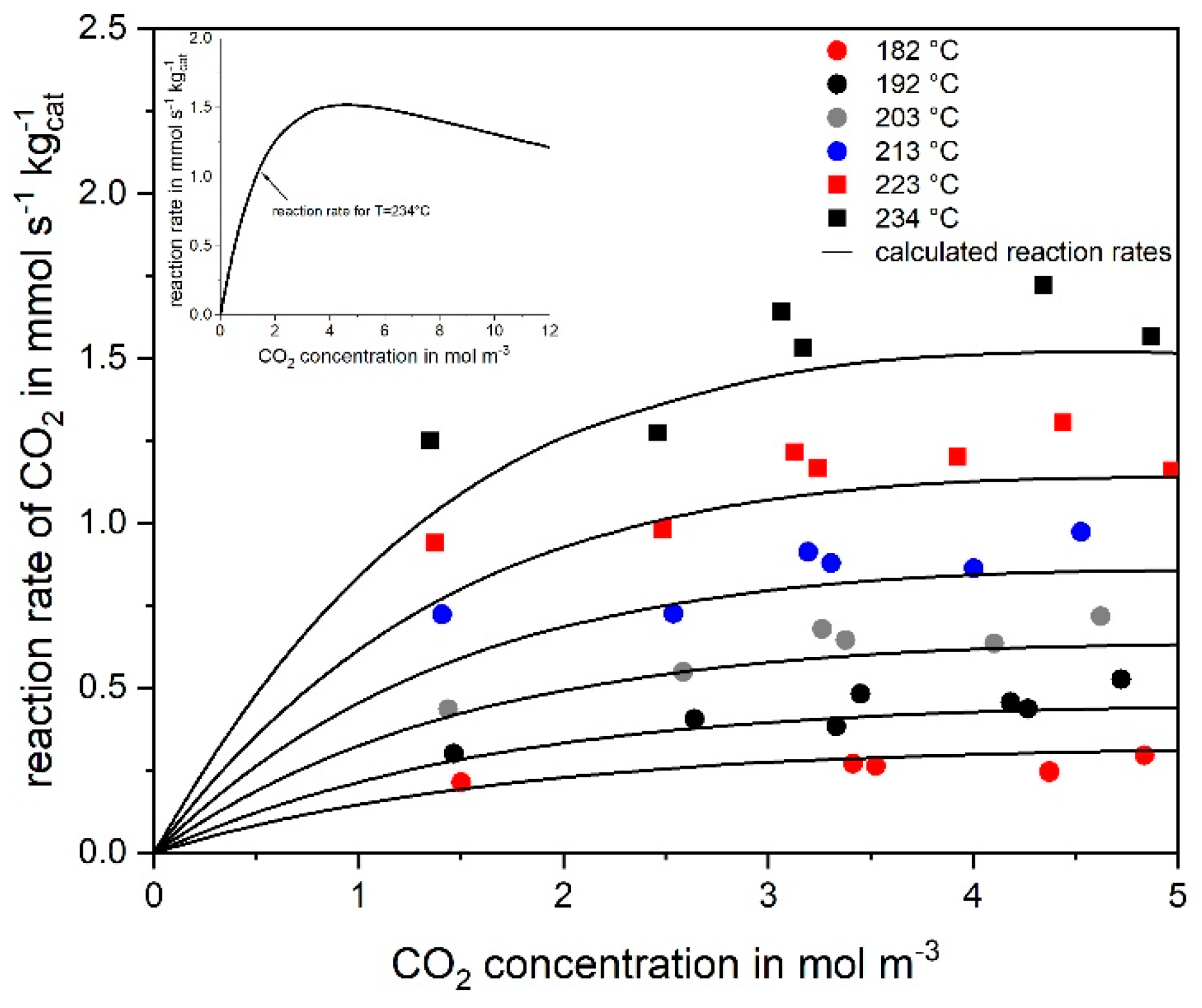
3.2. Kinetics of CO2 Methanation
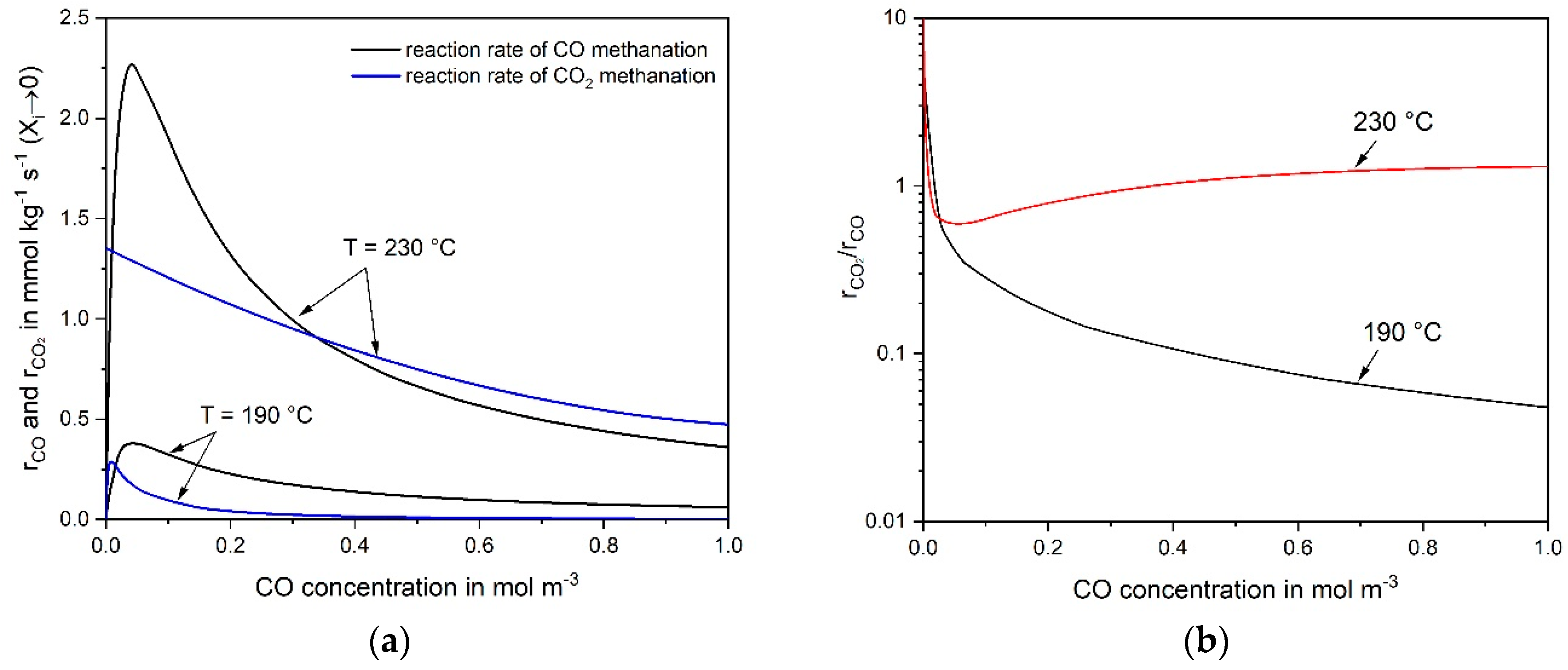
3.3. Reaction Mechanism of CO and CO2 Methanation
- The rate of RWGS can be estimated and compared with the measured CO2 rate based on the (easily) measurable rate of the reverse WGS reaction and some thermodynamic considerations.
- The rate of CO2 conversion must equal the rate of RWGS, if (hypothetically) only indirect CO2 methanation via CO takes place. The RWGS is then the rate determining step followed by fast CO methanation. A calculation based on the CO and CO2 rate equations (as determined in this work) lead to the concentration of the intermediate CO, which should correspond to the observed values. If the calculated CO yield is much higher than the one observed, this is an indication of the dominance of direct CO2 methanation and vice versa.
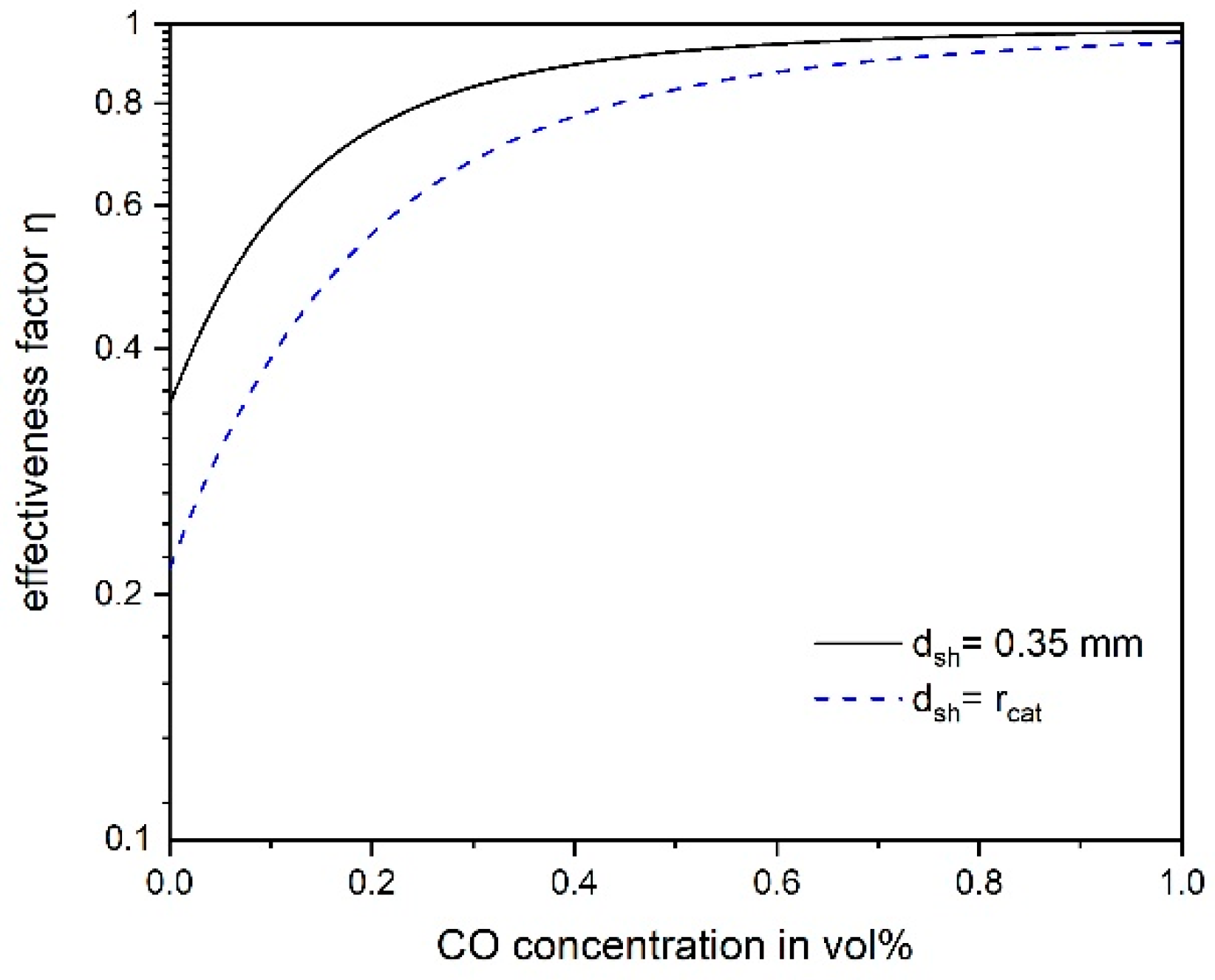
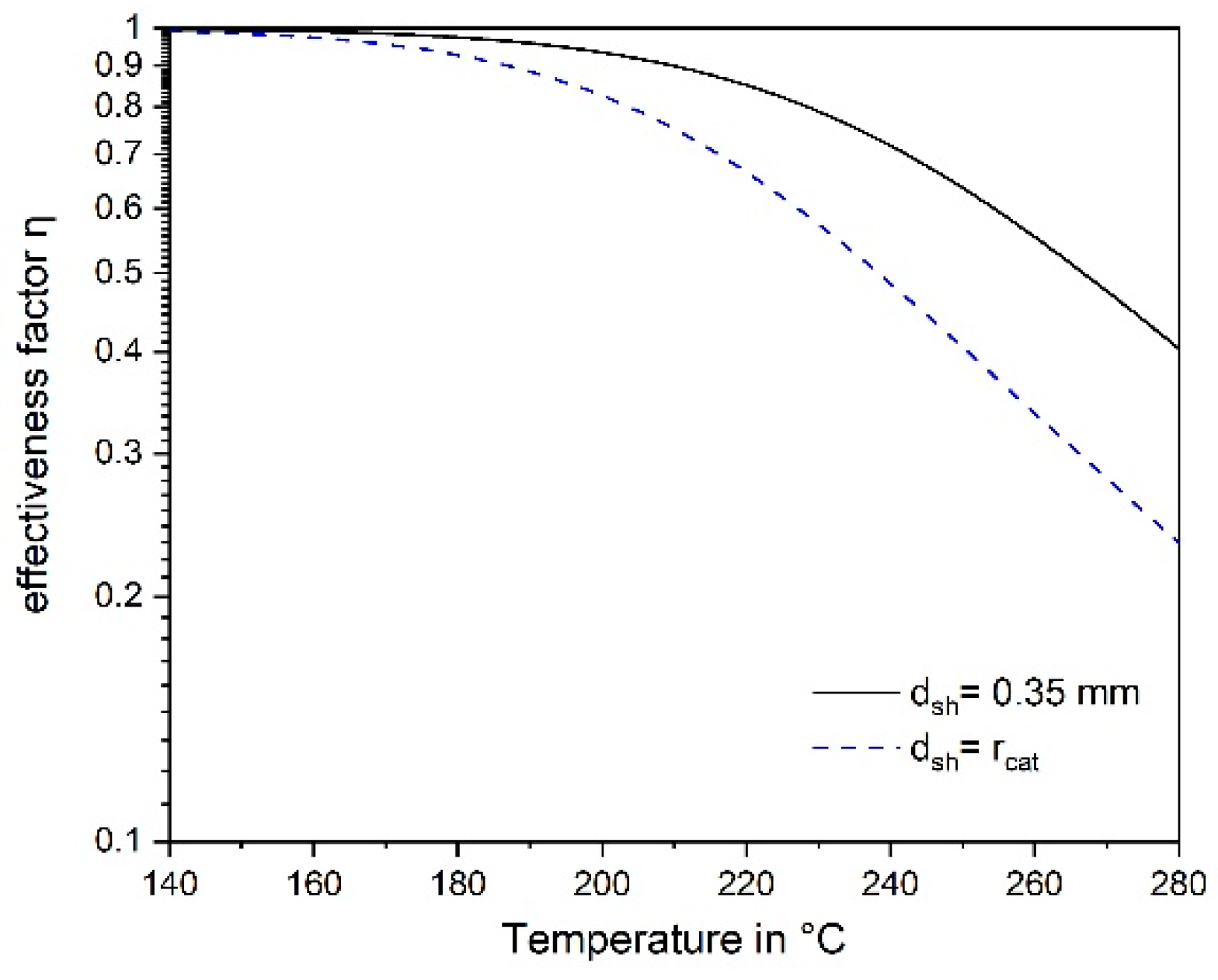
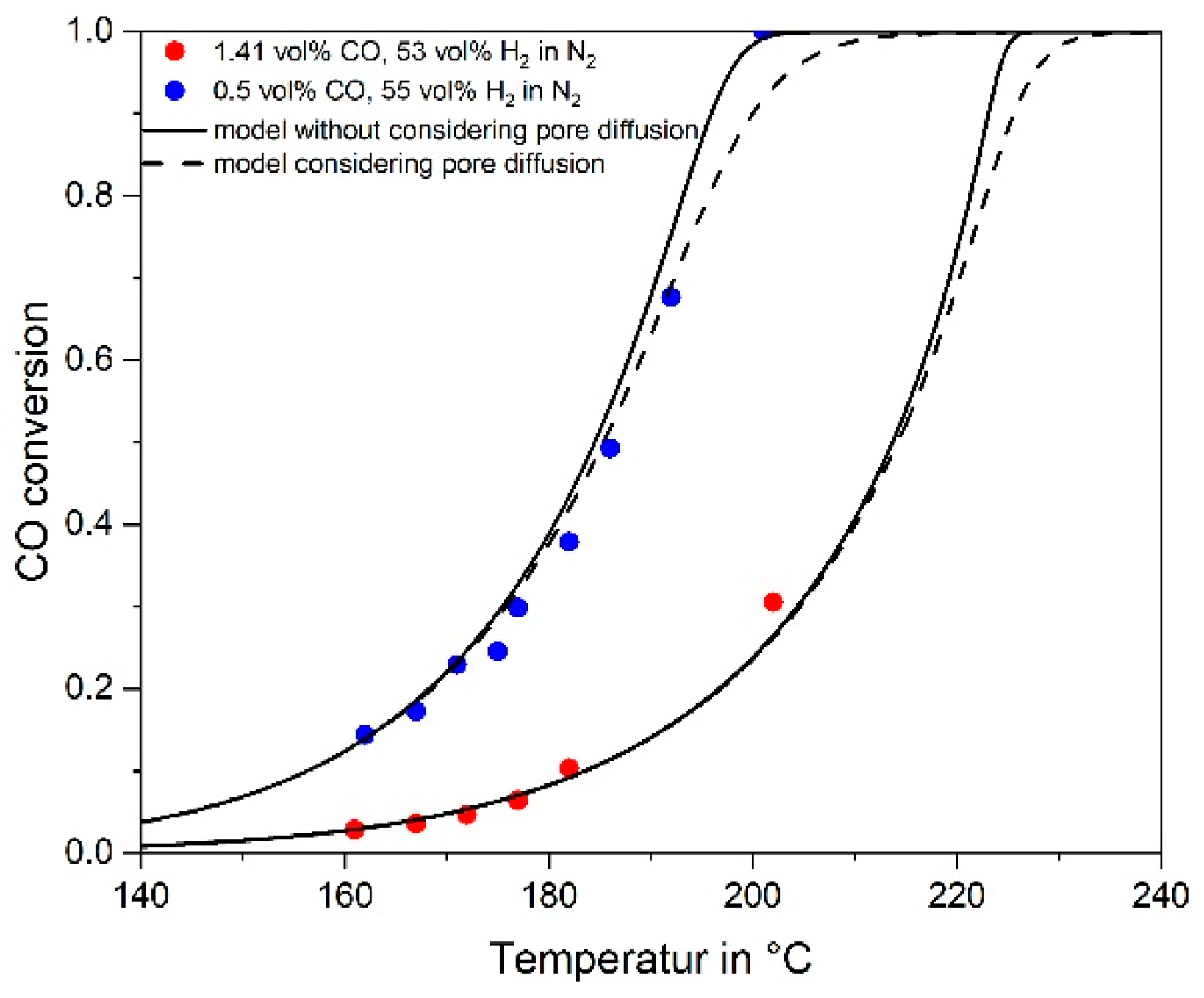
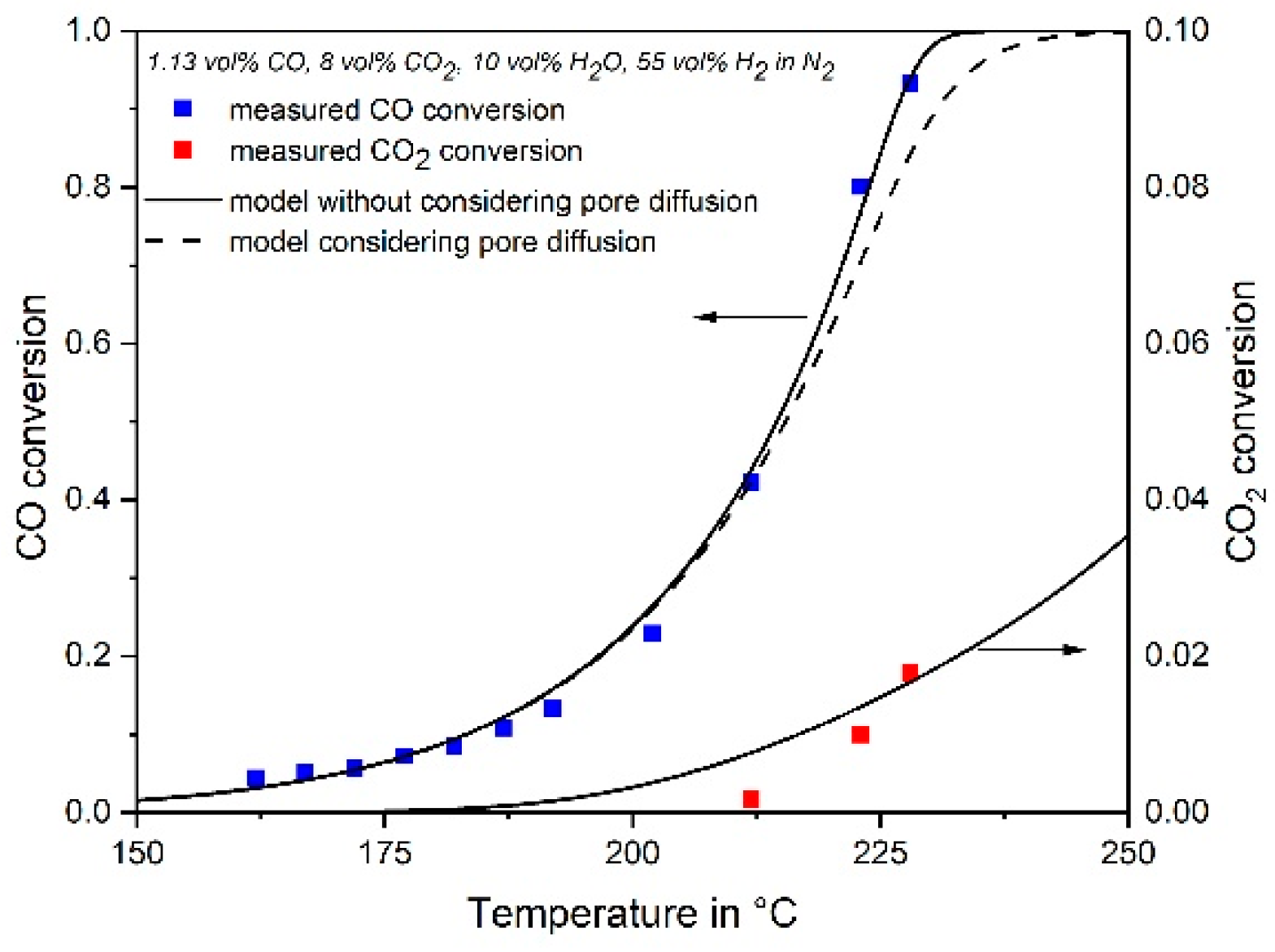
3.4. Influence of Pore Diffusion on the Rate of CO Methanation
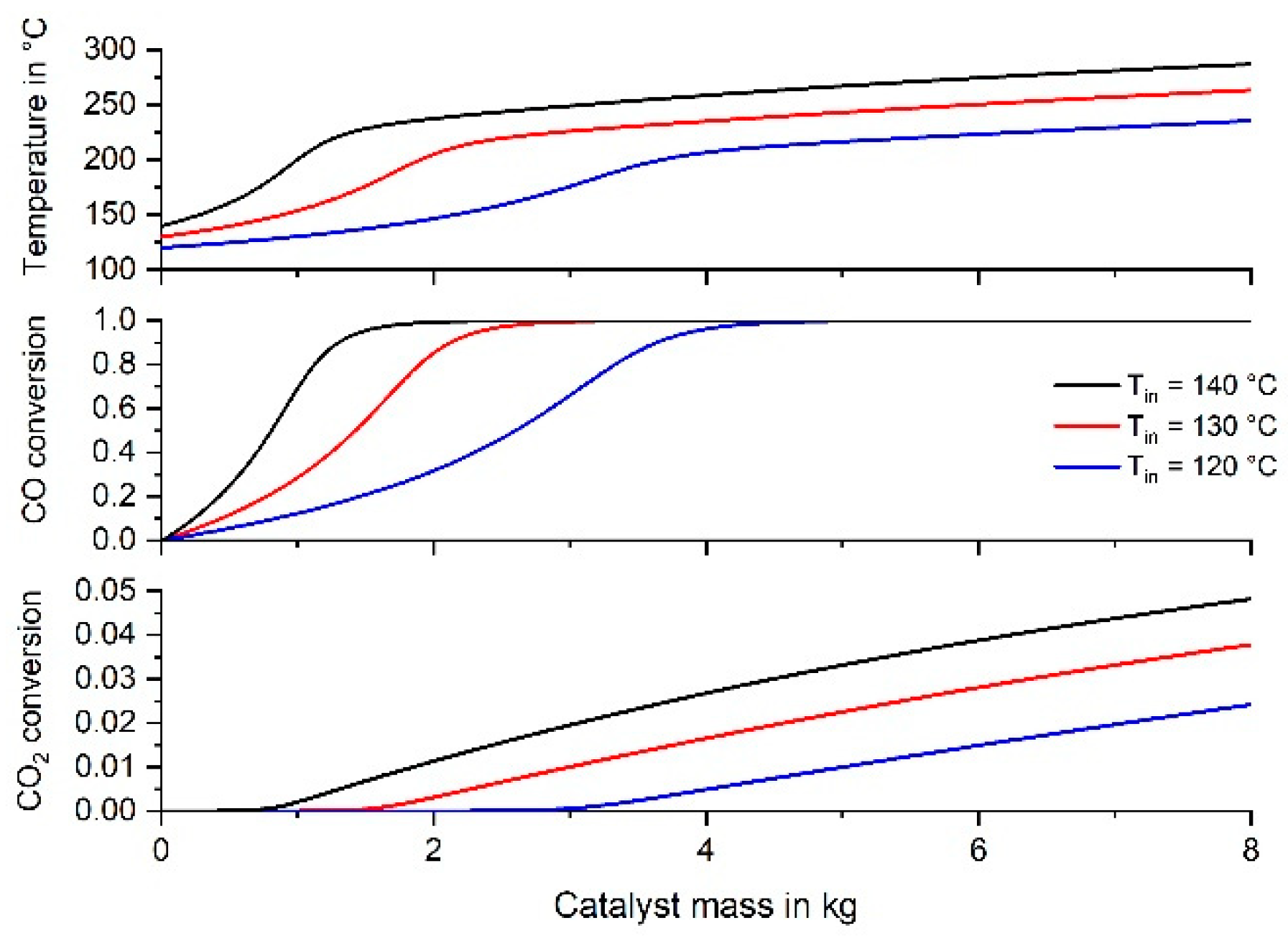
3.5. Simulation of CO Methanation in an Adiabatic Fixed Bed Reactor Suitable for a Household PEMFC
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eckle, S. Investigations of the Kinetics and Mechanism of the Selective Methanation of CO in CO2 and H2-Rich Reformates over Ru Supported Catalysts. Ph.D. Thesis, Verlag nicht Ermittelbar, Berlin, Germany, 2012. [Google Scholar]
- Djinović, P.; Galletti, C.; Specchia, S.; Specchia, V. Ru-based catalysts for CO selective methanation reaction in H2-rich gases. Catal. Today 2011, 164, 282–287. [Google Scholar] [CrossRef]
- Oh, S.H.; Sinkevitch, R.M. Carbon Monoxide Removal from Hydrogen-Rich Fuel Cell Feedstreams by Selective Catalytic Oxidation. J. Catal. 1993, 142, 254–262. [Google Scholar] [CrossRef]
- Peters, R.; Meißner, J. Gasaufbereitung für Brennstoffzellen. Chemie Ingenieur Technik 2004, 76, 1555–1558. [Google Scholar] [CrossRef]
- Hoogers, G. Fuel Cell Technology Handbook; CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Laguna, O.H.; Hernández, W.Y.; Arzamendi, G.; Gandía, L.M.; Centeno, M.A.; Odriozola, J.A. Gold supported on CuOx/CeO2 catalyst for the purification of hydrogen by the CO preferential oxidation reaction (PROX). Fuel 2014, 118, 176–185. [Google Scholar] [CrossRef]
- Mariño, F.; Baronetti, G.; Laborde, M.; Bion, N.; Le Valant, A.; Epron, F.; Duprez, D. Optimized CuO–CeO2 catalysts for COPROX reaction. Int. J. Hydrog. Energy 2008, 33, 1345–1353. [Google Scholar] [CrossRef]
- Reiche, A.; Haufe, S. Brennstoffzellen: Entwicklungsstand und Anwendungen. Chemie in Unserer Zeit 2004, 38, 400–411. [Google Scholar] [CrossRef]
- Galletti, C.; Specchia, S.; Saracco, G.; Specchia, V. CO-selective methanation over Ru–γAl2O3 catalysts in H2-rich gas for PEM FC applications. Chem. Eng. Sci. 2010, 65, 590–596. [Google Scholar] [CrossRef]
- Galletti, C.; Specchia, S.; Saracco, G.; Specchia, V. CO Methanation as Alternative Refinement Process for CO Abatement in H2-Rich Gas for PEM Applications. Int. J. Chem. React. Eng. 2007, 5. [Google Scholar] [CrossRef]
- Abdel-Mageed, A.M.; Widmann, D.; Olesen, S.E.; Chorkendorff, I.; Biskupek, J.; Behm, R.J. Selective CO Methanation on Ru/TiO2 Catalysts: Role and Influence of Metal–Support Interactions. ACS Catal. 2015, 5, 6753–6763. [Google Scholar] [CrossRef]
- Dagle, R.A.; Wang, Y.; Xia, G.-G.; Strohm, J.J.; Holladay, J.; Palo, D.R. Selective CO methanation catalysts for fuel processing applications. Appl. Catal. A Gener. 2007, 326, 213–218. [Google Scholar] [CrossRef]
- Urasaki, K.; Endo, K.-I.; Takahiro, T.; Kikuchi, R.; Kojima, T.; Satokawa, S. Effect of Support Materials on the Selective Methanation of CO over Ru Catalysts. Top. Catal. 2010, 53, 707–711. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Selective methanation of CO over supported noble metal catalysts: Effects of the nature of the metallic phase on catalytic performance. Appl. Catal. A Gener. 2008, 344, 45–54. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Selective methanation of CO over supported Ru catalysts. Appl. Catal. B Environ. 2009, 88, 470–478. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Mechanistic Study of the Selective Methanation of CO over Ru/TiO2 Catalyst: Identification of Active Surface Species and Reaction Pathways. J. Phys. Chem. C 2011, 115, 1220–1230. [Google Scholar] [CrossRef]
- Muñoz-Murillo, A.; Martínez, T.L.M.; Domínguez, M.I.; Odriozola, J.A.; Centeno, M.A. Selective CO methanation with structured RuO2/Al2O3 catalysts. Appl. Catal. B Environ. 2018, 236, 420–427. [Google Scholar] [CrossRef]
- Kowalczyk, Z.; Stołecki, K.; Raróg-Pilecka, W.; Miśkiewicz, E.; Wilczkowska, E.; Karpiński, Z. Supported ruthenium catalysts for selective methanation of carbon oxides at very low COx/H2 ratios. Appl. Catal. A Gener. 2008, 342, 35–39. [Google Scholar] [CrossRef]
- Garbis, P. Selective CO Methanation for PEMFC Applications. PhD Thesis, University of Bayreuth, Bayreuth, Germany, 2019. [Google Scholar]
- Kahlich, M.J.; Gasteiger, H.A.; Behm, R.J. Kinetics of the Selective Low-Temperature Oxidation of CO in H2-Rich Gas over Au/α-Fe2O3. J. Catal. 1999, 182, 430–440. [Google Scholar] [CrossRef]
- Goerke, O.; Pfeifer, P.; Schubert, K. Water gas shift reaction and selective oxidation of CO in microreactors. Appl. Catal. A Gener. 2004, 263, 11–18. [Google Scholar] [CrossRef]
- Echigo, M. A study of CO removal on an activated Ru catalyst for polymer electrolyte fuel cell applications. Appl. Catal. A Gener. 2003, 251, 157–166. [Google Scholar] [CrossRef]
- Choudhury, M.B.I.; Ahmed, S.; Shalabi, M.A.; Inui, T. Preferential methanation of CO in a syngas involving CO2 at lower temperature range. Appl. Catal. A 2006, 314, 47–53. [Google Scholar] [CrossRef]
- Ekerdt, J. Synthesis of hydrocarbons from CO and H2 over silica-supported Ru: Reaction rate measurements and infrared spectra of adsorbed species. J. Catal. 1979, 58, 170–187. [Google Scholar] [CrossRef]
- Underwood, R. The CO/H2 reaction over nickel-alumina studied by the transient method. J. Catal. 1984, 86, 245–253. [Google Scholar] [CrossRef]
- Prairie, M. A fourier transform infrared spectroscopic study of CO2 methanation on supported ruthenium. J. Catal. 1991, 129, 130–144. [Google Scholar] [CrossRef]
- Marwood, M.; van Vyve, F.; Doepper, R.; Renken, A. Periodic operation applied to the kinetic study of CO2 methanation. Catal. Today 1994, 20, 437–448. [Google Scholar] [CrossRef]
- Marwood, M.; Doepper, R.; Renken, A. In-situ surface and gas phase analysis for kinetic studies under transient conditions The catalytic hydrogenation of CO2. Appl. Catal. A Gener. 1997, 151, 223–246. [Google Scholar] [CrossRef]
- Falbo, L.; Martinelli, M.; Visconti, C.G.; Lietti, L.; Bassano, C.; Deiana, P. Kinetics of CO2 methanation on a Ru-based catalyst at process conditions relevant for Power-to-Gas applications. Appl. Catal. B Environ. 2018, 225, 354–363. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szanyi, J. CO2 Reduction on Supported Ru/Al2O3 Catalysts: Cluster Size Dependence of Product Selectivity. ACS Catal. 2013, 3, 2449–2455. [Google Scholar] [CrossRef]
- Mendes, D.; Mendes, A.; Madeira, L.M.; Iulianelli, A.; Sousa, J.M.; Basile, A. The water-gas shift reaction: From conventional catalytic systems to Pd-based membrane reactors—A review. Asia-Pac. J. Chem. Eng. 2010, 5, 111–137. [Google Scholar] [CrossRef]
- Levenspiel, O. The Chemical Reactor Omnibook; OSU Book Stores: Corvallis, OR, USA, 2002. [Google Scholar]
- Levenspiel, O. Chemical Reaction Engineering, 3rd ed.; Wiley: Hoboken, NJ, USA, 1999. [Google Scholar]
- Badenhop, T. Fuel Cells in the Energy Supply of Households. In Hydrogen and Fuel Cell; Töpler, J., Lehmann, J., Eds.; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2016; pp. 127–144. [Google Scholar]
- Viessmann. Brennstoffzellen-Heizgerät VITOVALOR PT2. Available online: https://www.viessmann.de/content/dam/vi-brands/DE/Produkte/Kraft-Waerme-Kopplung/Brennstoffzelle/Vitovalor-PT2/kpr-w-Vitovalor_PT2.pdf/_jcr_content/renditions/original.media_file.download_attachment.file/kpr-w-Vitovalor_PT2.pdf (accessed on 17 December 2018).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| Particle radius, rp | 1.25 mm |
| Shell thickness, dsh | 0.35 mm |
| Density of particle, ρp | 980 kg·m−3 |
| Porosity of particle, εp | 0.49 |
| Tortuosity, τp | 2 |
| Average pore diameter, dpore | 9.3 nm |
| BET surface area | 215 m2·g−1 |
| Ru particle size | 6 nm |
| Parameter | Value |
|---|---|
| EA,CO | 90 kJ·mol−1 |
| k0,CO | 3.61 × 107 m6·s−1·kg−1·mol−1 |
| K1 | 23 m3·mol−1 |
| K2 | 0.3 m3·mol−1 |
| Parameter | Value |
|---|---|
| EA,CO2 | 81 kJ·mol−1 |
| k0,CO2 | 2.8 ×104 m6·s−1·kg−1·mol−1 |
| K0,3 | 8.4 m3·mol−1 |
| ΔH3 | 15 kJ·mol−1 |
| K0,4 | 1.15 ×10−13 m3·mol−1 |
| ΔH4 | −125 kJ·mol−1 |
| K5 | 1.1 m3·mol−1 |
| Tin in °C | Tout in °C | mcat in kg | XCO in % | XCO2 in % | XH2 in % | EH2 | CO Content at Reactor Outlet |
|---|---|---|---|---|---|---|---|
| 120 | 210 | 4.25 | 98.00 | 0.61 | 2.16 | 1.17 | Limit value of 100 ppm just reached |
| 225 | 6.40 | 99.99 | 1.68 | 2.73 | 1.48 | <1 ppm | |
| 238 | 8.50 | 99.99 | 2.60 | 3.19 | 1.73 | <1 ppm | |
| 130 | 221 | 2.60 | 98.00 | 0.74 | 2.22 | 1.20 | Limit value of 100 ppm just reached |
| 234 | 3.90 | 99.95 | 1.59 | 2.69 | 1.45 | 3 ppm | |
| 245 | 5.20 | 99.99 | 2.37 | 3.08 | 1.67 | <1 ppm | |
| 140 | 233 | 1.70 | 98.00 | 0.90 | 2.30 | 1.24 | Limit value of 100 ppm just reached |
| 244 | 2.55 | 99.86 | 1.60 | 2.69 | 1.45 | 7 ppm | |
| 253 | 3.40 | 99.99 | 2.25 | 3.02 | 1.63 | <1 ppm |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garbis, P.; Kern, C.; Jess, A. Kinetics and Reactor Design Aspects of Selective Methanation of CO over a Ru/γ-Al2O3 Catalyst in CO2/H2 Rich Gases. Energies 2019, 12, 469. https://doi.org/10.3390/en12030469
Garbis P, Kern C, Jess A. Kinetics and Reactor Design Aspects of Selective Methanation of CO over a Ru/γ-Al2O3 Catalyst in CO2/H2 Rich Gases. Energies. 2019; 12(3):469. https://doi.org/10.3390/en12030469
Chicago/Turabian StyleGarbis, Panagiota, Christoph Kern, and Andreas Jess. 2019. "Kinetics and Reactor Design Aspects of Selective Methanation of CO over a Ru/γ-Al2O3 Catalyst in CO2/H2 Rich Gases" Energies 12, no. 3: 469. https://doi.org/10.3390/en12030469