Current Evidence for Immune Checkpoint Inhibition in Advanced Hepatocellular Carcinoma

 , , and
, , and
Abstract
:
1. Introduction
2. Systemic Therapies in aHCC
3. ICI in HCC
3.1. Single-Agent ICI
3.1.1. CTLA-4 Inhibitors
3.1.2. PD-1/PD-L1 Inhibitors
3.2. Dual Checkpoint Inhibition
3.3. ICI/VEGF Inhibition
4. Intermediate Stage Disease
5. Evaluation of Predictive Biomarkers for ICI
5.1. PD-L1 Expression
5.2. Tumour Mutation Burden (TMB)
5.3. Microsatellite Instability (MSI)
5.4. Immune Cell Infiltration/Tumour Microenvironment
5.5. Viral Aetiology and Response to ICI
5.6. ICI in aHCC and Impaired Liver Function
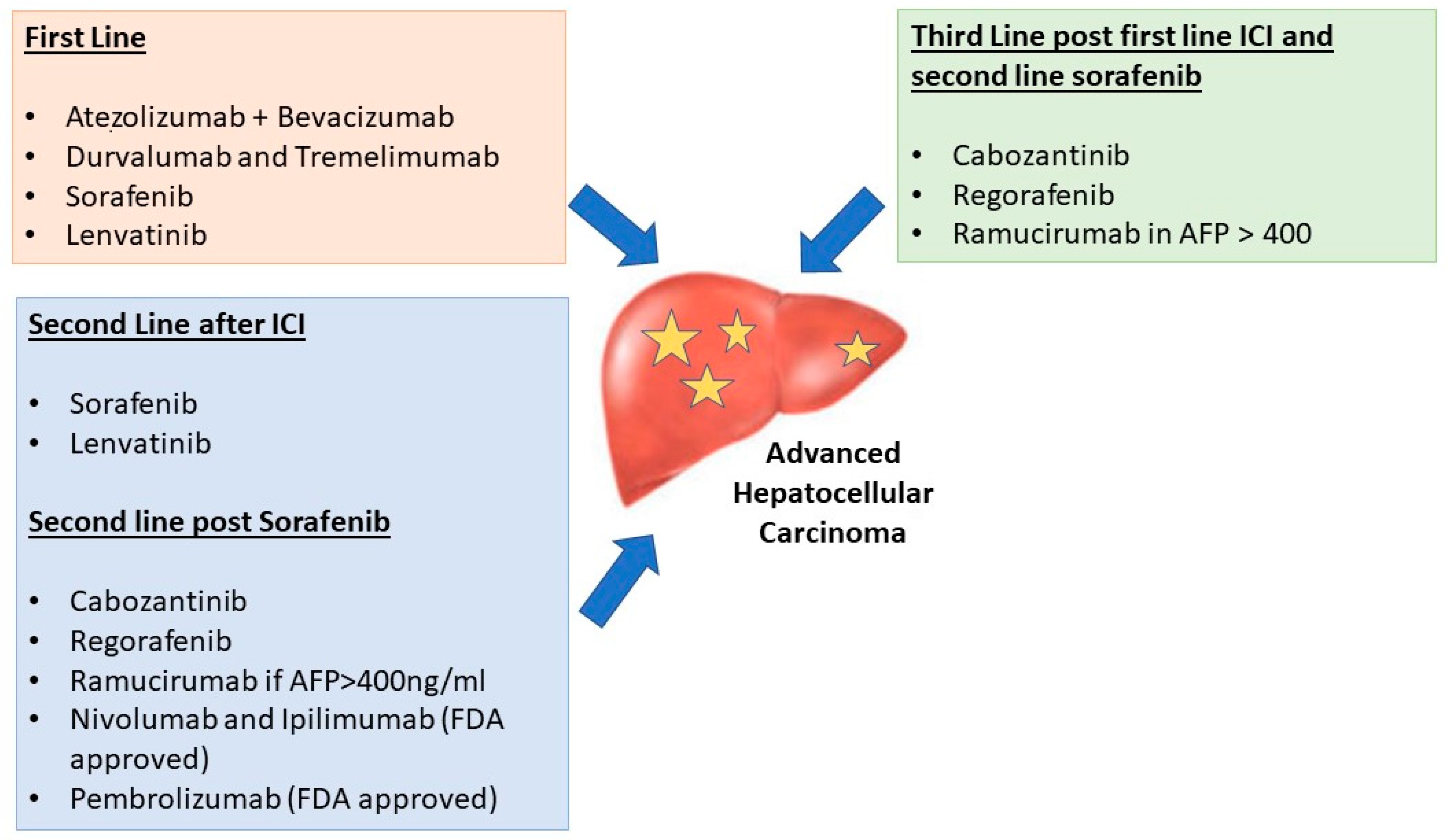
6. Selection of First-Line Therapy in aHCC
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular Carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; Terrault, N.A.; Tacke, F.; Gluud, L.L.; Arrese, M.; Bugianesi, E.; Loomba, R. Global Epidemiology of Cirrhosis—Aetiology, Trends and Predictions. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 388–398. [Google Scholar] [CrossRef]
- Dasgupta, P.; Henshaw, C.; Youlden, D.R.; Clark, P.J.; Aitken, J.F.; Baade, P.D. Global Trends in Incidence Rates of Primary Adult Liver Cancers: A Systematic Review and Meta-Analysis. Front. Oncol. 2020, 10, 171. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Allemani, C.; Matsuda, T.; Di Carlo, V.; Harewood, R.; Matz, M.; Nikšić, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Estève, J.; et al. Global Surveillance of Trends in Cancer Survival 2000-14 (CONCORD-3): Analysis of Individual Records for 37 513 025 Patients Diagnosed with One of 18 Cancers from 322 Population-Based Registries in 71 Countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef] [PubMed]
- Rumgay, H.; Arnold, M.; Ferlay, J.; Lesi, O.; Cabasag, C.J.; Vignat, J.; Laversanne, M.; McGlynn, K.A.; Soerjomataram, I. Global Burden of Primary Liver Cancer in 2020 and Predictions to 2040. J. Hepatol. 2022, 77, 1598–1606. [Google Scholar] [CrossRef]
- Llovet, J.M.; Brú, C.; Bruix, J. Prognosis of Hepatocellular Carcinoma: The BCLC Staging Classification. Semin. Liver Dis. 1999, 19, 329–338. [Google Scholar] [CrossRef]
- Galle, P.R.; Forner, A.; Llovet, J.M.; Mazzaferro, V.; Piscaglia, F.; Raoul, J.-L.; Schirmacher, P.; Vilgrain, V. EASL Clinical Practice Guidelines: Management of Hepatocellular Carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef]
- Lohitesh, K.; Chowdhury, R.; Mukherjee, S. Resistance a Major Hindrance to Chemotherapy in Hepatocellular Carcinoma: An Insight. Cancer Cell Int. 2018, 18, 44. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- da Fonseca, L.G.; Reig, M.; Bruix, J. Tyrosine Kinase Inhibitors and Hepatocellular Carcinoma. Clin. Liver Dis. 2020, 24, 719–737. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J.G.; Macias, R.I.R.; Monte, M.J.; Romero, M.R.; Asensio, M.; Sanchez-Martin, A.; Cives-Losada, C.; Temprano, A.G.; Espinosa-Escudero, R.; Reviejo, M.; et al. Molecular Bases of Drug Resistance in Hepatocellular Carcinoma. Cancers 2020, 12, 1663. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X. Systemic Therapy of Advanced Hepatocellular Carcinoma: How Hopeful Should We Be? Oncologist 2006, 11, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.-L.; Guan, Z.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Yang, T.-S.; Tak, W.Y.; Pan, H.; Yu, S.; et al. Efficacy and Safety of Sorafenib in Patients with Advanced Hepatocellular Carcinoma According to Baseline Status: Subset Analyses of the Phase III Sorafenib Asia–Pacific Trial. Eur. J. Cancer 2012, 48, 1452–1465. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus Sorafenib in First-Line Treatment of Patients with Unresectable Hepatocellular Carcinoma: A Randomised Phase 3 Non-Inferiority Trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for Patients with Hepatocellular Carcinoma Who Progressed on Sorafenib Treatment (RESORCE): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after Sorafenib in Patients with Advanced Hepatocellular Carcinoma and Increased α-Fetoprotein Concentrations (REACH-2): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Tian, Y.; Lei, Y.; Fu, Y.; Sun, H.; Wang, J.; Xia, F. Molecular Mechanisms of Resistance to Tyrosine Kinase Inhibitors Associated with Hepatocellular Carcinoma. Curr. Cancer Drug Targets 2022, 22, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B. Mechanisms of Resistance to Sorafenib and the Corresponding Strategies in Hepatocellular Carcinoma. World J. Hepatol. 2013, 5, 345. [Google Scholar] [CrossRef] [PubMed]
- Rimassa, L.; Danesi, R.; Pressiani, T.; Merle, P. Management of Adverse Events Associated with Tyrosine Kinase Inhibitors: Improving Outcomes for Patients with Hepatocellular Carcinoma. Cancer Treat. Rev. 2019, 77, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, W.K.; Rumble, R.B.; Van Veldhuizen, P.J.; Al-Ahmadie, H.; Emamekhoo, H.; Hauke, R.J.; Louie, A.V.; Milowsky, M.I.; Molina, A.M.; Rose, T.L.; et al. Management of Metastatic Clear Cell Renal Cell Carcinoma: ASCO Guideline. J. Clin. Oncol. 2022, 40, 2957–2995. [Google Scholar] [CrossRef] [PubMed]
- Govindan, R.; Aggarwal, C.; Antonia, S.J.; Davies, M.; Dubinett, S.M.; Ferris, A.; Forde, P.M.; Garon, E.B.; Goldberg, S.B.; Hassan, R.; et al. Society for Immunotherapy of Cancer (SITC) Clinical Practice Guideline on Immunotherapy for the Treatment of Lung Cancer and Mesothelioma. J. Immunother. Cancer 2022, 10, e003956. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A Decade of Immune-Checkpoint Inhibitors in Cancer Therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Switzer, B.; Puzanov, I.; Skitzki, J.J.; Hamad, L.; Ernstoff, M.S. Managing Metastatic Melanoma in 2022: A Clinical Review. JCO Oncol. Pract. 2022, 18, 335–351. [Google Scholar] [CrossRef]
- Borcoman, E.; Kanjanapan, Y.; Champiat, S.; Kato, S.; Servois, V.; Kurzrock, R.; Goel, S.; Bedard, P.; Le Tourneau, C. Novel Patterns of Response under Immunotherapy. Ann. Oncol. 2019, 30, 385–396. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive Strategies That Are Mediated by Tumor Cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver Immunology and Its Role in Inflammation and Homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Keenan, B.P.; Fong, L.; Kelley, R.K. Immunotherapy in Hepatocellular Carcinoma: The Complex Interface between Inflammation, Fibrosis, and the Immune Response. J. Immunother. Cancer 2019, 7, 267. [Google Scholar] [CrossRef] [PubMed]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Iñarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A Clinical Trial of CTLA-4 Blockade with Tremelimumab in Patients with Hepatocellular Carcinoma and Chronic Hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in Patients with Advanced Hepatocellular Carcinoma (CheckMate 040): An Open-Label, Non-Comparative, Phase 1/2 Dose Escalation and Expansion Trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Matilla, A.; Santoro, A.; Melero, I.; Gracián, A.C.; Acosta-Rivera, M.; Choo, S.-P.; El-Khoueiry, A.B.; Kuromatsu, R.; El-Rayes, B.; et al. CheckMate 040 Cohort 5: A Phase I/II Study of Nivolumab in Patients with Advanced Hepatocellular Carcinoma and Child-Pugh B Cirrhosis. J. Hepatol. 2021, 75, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.H.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Updated Efficacy and Safety of KEYNOTE-224: A Phase II Study of Pembrolizumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib. Eur. J. Cancer 2022, 167, 1–12. [Google Scholar] [CrossRef]
- Verset, G.; Borbath, I.; Karwal, M.; Verslype, C.; Van Vlierberghe, H.; Kardosh, A.; Zagonel, V.; Stal, P.; Sarker, D.; Palmer, D.H.; et al. Pembrolizumab Monotherapy for Previously Untreated Advanced Hepatocellular Carcinoma: Data from the Open-Label, Phase II KEYNOTE-224 Trial. Clin. Cancer Res. 2022, 28, 2547–2554. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.-W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus Sorafenib in Advanced Hepatocellular Carcinoma (CheckMate 459): A Randomised, Multicentre, Open-Label, Phase 3 Trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef]
- Qin, S.; Finn, R.S.; Kudo, M.; Meyer, T.; Vogel, A.; Ducreux, M.; Macarulla, T.M.; Tomasello, G.; Boisserie, F.; Hou, J.; et al. RATIONALE 301 Study: Tislelizumab versus Sorafenib as First-Line Treatment for Unresectable Hepatocellular Carcinoma. Future Oncol. 2019, 15, 1811–1822. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chan, S.L.; Kudo, M.; Lau, G.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Dao, T.V.; De Toni, E.N.; et al. Phase 3 Randomized, Open-Label, Multicenter Study of Tremelimumab (T) and Durvalumab (D) as First-Line Therapy in Patients (Pts) with Unresectable Hepatocellular Carcinoma (UHCC): HIMALAYA. J. Clin. Oncol. 2022, 40, 379. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated Efficacy and Safety Data from IMbrave150: Atezolizumab plus Bevacizumab vs. Sorafenib for Unresectable Hepatocellular Carcinoma. J. Hepatol. 2022, 76, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Xu, J.; Bai, Y.; Xu, A.; Cang, S.; Du, C.; Li, Q.; Lu, Y.; Chen, Y.; Guo, Y.; et al. Sintilimab plus a Bevacizumab Biosimilar (IBI305) versus Sorafenib in Unresectable Hepatocellular Carcinoma (ORIENT-32): A Randomised, Open-Label, Phase 2-3 Study. Lancet Oncol. 2021, 22, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Rimassa, L.; Cheng, A.-L.; Kaseb, A.; Qin, S.; Zhu, A.X.; Chan, S.L.; Melkadze, T.; Sukeepaisarnjaroen, W.; Breder, V.; et al. Cabozantinib plus Atezolizumab versus Sorafenib for Advanced Hepatocellular Carcinoma (COSMIC-312): A Multicentre, Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2022, 23, 995–1008. [Google Scholar] [CrossRef]
- Qin, S.; Chan, S.L.; Gu, S.; Bai, Y.; Ren, Z.; Lin, X.; Chen, Z.; Jia, W.; Jin, Y.; Guo, Y.; et al. Camrelizumab plus Rivoceranib versus Sorafenib as First-Line Therapy for Unresectable Hepatocellular Carcinoma (CARES-310): A Randomised, Open-Label, International Phase 3 Study. Lancet 2023. [Google Scholar] [CrossRef]
- Finn, R.S.; Kudo, M.; Merle, P.; Meyer, T.; Qin, S.; Ikeda, M.; Xu, R.; Edeline, J.; Ryoo, B.-Y.; Ren, Z.; et al. LBA34 Primary Results from the Phase III LEAP-002 Study: Lenvatinib plus Pembrolizumab versus Lenvatinib as First-Line (1L) Therapy for Advanced Hepatocellular Carcinoma (AHCC). Ann. Oncol. 2022, 33, S1401. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients with Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Qin, S.; Chen, Z.; Fang, W.; Ren, Z.; Xu, R.; Ryoo, B.-Y.; Meng, Z.; Bai, Y.; Chen, X.; Liu, X.; et al. Pembrolizumab plus Best Supportive Care versus Placebo plus Best Supportive Care as Second-Line Therapy in Patients in Asia with Advanced Hepatocellular Carcinoma (HCC): Phase 3 KEYNOTE-394 Study. J. Clin. Oncol. 2022, 40, 383. [Google Scholar] [CrossRef]
- Sangro, B.; Park, J.; Finn, R.; Cheng, A.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.; Harding, J.; Merle, P.; et al. LBA-3 CheckMate 459: Long-Term (Minimum Follow-up 33.6 Months) Survival Outcomes with Nivolumab versus Sorafenib as First-Line Treatment in Patients with Advanced Hepatocellular Carcinoma. Ann. Oncol. 2020, 31, S241–S242. [Google Scholar] [CrossRef]
- Squibb BM Bristol Myers Squibb Statement on Opdivo® (Nivolumab) Monotherapy Post-Sorafenib Hepatocellular Carcinoma U.S. Indication 2021. Available online: https://news.bms.com/news/corporate-financial/2021/Bristol-Myers-Squibb-Statement-on-Opdivo-nivolumab-Monotherapy-Post-Sorafenib-Hepatocellular-Carcinoma-U.S.-Indication/default.aspx (accessed on 30 June 2023).
- Kudo, M.; Lim, H.Y.; Cheng, A.-L.; Chao, Y.; Yau, T.; Ogasawara, S.; Kurosaki, M.; Morimoto, N.; Ohkawa, K.; Yamashita, T.; et al. Pembrolizumab as Second-Line Therapy for Advanced Hepatocellular Carcinoma: A Subgroup Analysis of Asian Patients in the Phase 3 KEYNOTE-240 Trial. Liver Cancer 2021, 10, 275–284. [Google Scholar] [CrossRef]
- Finn, R.S.; Gu, K.; Chen, X.; Merle, P.; Lee, K.-H.; Bouattour, M.; Cao, P.; Wang, W.; Cheng, A.-L.; Zhu, L.; et al. Abstract CT222: Pembrolizumab (Pembro) for Previously Treated Advanced Hepatocellular Carcinoma (AHCC): Meta-Analysis of the Phase 3 KEYNOTE-240 and KEYNOTE-394 Studies. Cancer Res. 2022, 82, CT222. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Kudo, M.; Meyer, T.; Boisserie, F.; Li, S.; Chen, Y.; Barnes, G.; Abdrashitov, R.; Zhu, A.X.; et al. Tislelizumab versus Sorafenib in First-Line Treatment of Unresectable Hepatocellular Carcinoma: Impact on Health-Related Quality of Life in RATIONALE-301 Population. J. Clin. Oncol. 2023, 41, 495. [Google Scholar] [CrossRef]
- Qin, S.; Ren, Z.; Meng, Z.; Chen, Z.; Chai, X.; Xiong, J.; Bai, Y.; Yang, L.; Zhu, H.; Fang, W.; et al. Camrelizumab in Patients with Previously Treated Advanced Hepatocellular Carcinoma: A Multicentre, Open-Label, Parallel-Group, Randomised, Phase 2 Trial. Lancet Oncol. 2020, 21, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Yau, T.; Kang, Y.-K.; Kim, T.-Y.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Nivolumab (NIVO) plus Ipilimumab (IPI) Combination Therapy in Patients (Pts) with Advanced Hepatocellular Carcinoma (AHCC): Long-Term Results from CheckMate 040. J. Clin. Oncol. 2021, 39, 269. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Morse, M.A.; Sun, W.; Kim, R.; He, A.R.; Abada, P.B.; Mynderse, M.; Finn, R.S. The Role of Angiogenesis in Hepatocellular Carcinoma. Clin. Cancer Res. 2019, 25, 912–920. [Google Scholar] [CrossRef]
- Hegde, P.S.; Wallin, J.J.; Mancao, C. Predictive Markers of Anti-VEGF and Emerging Role of Angiogenesis Inhibitors as Immunotherapeutics. Semin. Cancer Biol. 2018, 52, 117–124. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Sun, M.; Xu, A.; Bai, Y.; Wu, J.; Shao, G.; Song, L.; Jin, X.; Song, W.; Li, B.; et al. Phase 2 Study of AK104 (PD-1/CTLA-4 Bispecific Antibody) plus Lenvatinib as First-Line Treatment of Unresectable Hepatocellular Carcinoma. J. Clin. Oncol. 2021, 39, 4101. [Google Scholar] [CrossRef]
- Hsieh, D.; Kainthla, R.; Zhu, H.; Beg, M.S. Phase 2 Trial of Pembrolizumab (Pembro) and Bavituximab (Bavi) in Advanced Hepatocellular Carcinoma (HCC). J. Clin. Oncol. 2023, 41, 584. [Google Scholar] [CrossRef]
- Yoo, C.; Ryoo, B.-Y.; Kim, H.-D.; Ryu, M.-H.; Kang, B.; Chon, H.J.; Hong, J.Y.; Lim, H.Y. Regorafenib plus Nivolumab as First-Line Therapy for Unresectable Hepatocellular Carcinoma (UHCC): Multicenter Phase 2 Trial (RENOBATE). J. Clin. Oncol. 2022, 40, 415. [Google Scholar] [CrossRef]
- Zhang, F.; Bai, Y.; Fang, W.; Meng, Z.; Xiong, J.; Guo, Y.; Zhang, T.; Zhang, J.; Ying, J.; Chen, Z.; et al. Safety, Tolerability, and Preliminary Antitumor Activity of Sitravatinib plus Tislelizumab (TIS) in Patients (Pts) with Unresectable Locally Advanced or Metastatic Hepatocellular Carcinoma (HCC). J. Clin. Oncol. 2022, 40, 418. [Google Scholar] [CrossRef]
- Vogel, A.; Siegler, G.M.; Siebler, J.; Lindig, U.; Schultheiß, M.; Müller, T.; Simon, H.; Jöckel, C.; Mueller, D.W.; Al-Batran, S.-E.; et al. IMMUNIB Trial (AIO-HEP-0218/Ass): A Single-Arm, Phase II Study Evaluating Safety and Efficacy of Immunotherapy Nivolumab in Combination with Lenvatinib in Advanced-Stage Hepatocellular Carcinoma (HCC). J. Clin. Oncol. 2022, 40, 4107. [Google Scholar] [CrossRef]
- Sanduzzi Zamparelli, M.; Matilla, A.; Lledó, J.L.; Martínez, S.M.; Varela, M.; Iñarrairaegui, M.; Perelló, C.; Minguez, B.; Llarch, N.; Márquez, L.; et al. Early Nivolumab Addition to Regorafenib in Patients with Hepatocellular Carcinoma Progressing under First-Line Therapy (GOING Trial), Interim Analysis and Safety Profile. J. Clin. Oncol. 2022, 40, 428. [Google Scholar] [CrossRef]
- Kudo, M.; Motomura, K.; Wada, Y.; Inaba, Y.; Sakamoto, Y.; Kurosaki, M.; Umeyama, Y.; Kamei, Y.; Yoshimitsu, J.; Fujii, Y.; et al. Avelumab in Combination with Axitinib as First-Line Treatment in Patients with Advanced Hepatocellular Carcinoma: Results from the Phase 1b VEGF Liver 100 Trial. Liver Cancer 2021, 10, 249–259. [Google Scholar] [CrossRef]
- Yau, T.; Zagonel, V.; Santoro, A.; Acosta-Rivera, M.; Choo, S.P.; Matilla, A.; He, A.R.; Cubillo Gracian, A.; El-Khoueiry, A.B.; Sangro, B.; et al. Nivolumab Plus Cabozantinib With or Without Ipilimumab for Advanced Hepatocellular Carcinoma: Results from Cohort 6 of the CheckMate 040 Trial. J. Clin. Oncol. 2023, 41, 1747–1757. [Google Scholar] [CrossRef]
- Guo, L.Y.; Zhu, P.; Jin, X.P. Association between the Expression of HIF-1α and VEGF and Prognostic Implications in Primary Liver Cancer. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef]
- Mizukoshi, E.; Yamashita, T.; Arai, K.; Sunagozaka, H.; Ueda, T.; Arihara, F.; Kagaya, T.; Yamashita, T.; Fushimi, K.; Kaneko, S. Enhancement of Tumor-Associated Antigen-Specific T Cell Responses by Radiofrequency Ablation of Hepatocellular Carcinoma. Hepatology 2013, 57, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Leuchte, K.; Staib, E.; Thelen, M.; Gödel, P.; Lechner, A.; Zentis, P.; Garcia-Marquez, M.; Waldschmidt, D.; Datta, R.R.; Wahba, R.; et al. Microwave Ablation Enhances Tumor-Specific Immune Response in Patients with Hepatocellular Carcinoma. Cancer Immunol. Immunother. 2021, 70, 893–907. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in Combination with Ablation in Patients with Advanced Hepatocellular Carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Buchalter, J.; Browne, I.; Mac Eochagain, C.; Flynn, C.; Carroll, H.K.; Galligan, M.; Bourke, M.; Lester-Grant, A.; Desmond, F.; Hoey, K.; et al. Tremelimumab (Day 1 Only) and Durvalumab in Combination with Transarterial Chemoemobilization (TACE) in Patients with Hepatocellular Carcinoma (HCC). J. Clin. Oncol. 2022, 40, 451. [Google Scholar] [CrossRef]
- Sangro, B.; Kudo, M.; Qin, S.; Ren, Z.; Chan, S.; Joseph, E.; Arai, Y.; Mann, H.; Morgan, S.; Cohen, G.; et al. P-347 A Phase 3, Randomized, Double-Blind, Placebo-Controlled Study of Transarterial Chemoembolization Combined with Durvalumab or Durvalumab plus Bevacizumab Therapy in Patients with Locoregional Hepatocellular Carcinoma: EMERALD-1. Ann. Oncol. 2020, 31, S202–S203. [Google Scholar] [CrossRef]
- Sangro, B.; Harding, J.J.; Johnson, M.; Palmer, D.H.; Edeline, J.; Abou-Alfa, G.K.; Cheng, A.-L.; Decaens, T.; El-Khoueiry, A.B.; Finn, R.S.; et al. A Phase III, Double-Blind, Randomized Study of Nivolumab (NIVO) and Ipilimumab (IPI), Nivo Monotherapy or Placebo plus Transarterial Chemoembolization (TACE) in Patients with Intermediate-Stage Hepatocellular Carcinoma (HCC). J. Clin. Oncol. 2021, 39, TPS349. [Google Scholar] [CrossRef]
- Llovet, J.M.; Vogel, A.; Madoff, D.C.; Finn, R.S.; Ogasawara, S.; Ren, Z.; Mody, K.; Li, J.J.; Siegel, A.B.; Dubrovsky, L.; et al. Randomized Phase 3 LEAP-012 Study: Transarterial Chemoembolization with or Without Lenvatinib Plus Pembrolizumab for Intermediate-Stage Hepatocellular Carcinoma Not Amenable to Curative Treatment. Cardiovasc. Intervent Radiol. 2022, 45, 405–412. [Google Scholar] [CrossRef]
- Kloeckner, R.; Galle, P.R.; Bruix, J. Local and Regional Therapies for Hepatocellular Carcinoma. Hepatology 2021, 73, 137–149. [Google Scholar] [CrossRef]
- Kudo, M.; Guo, Y.; Hua, Y.; Zhao, M.; Xing, W.; Zhang, Y.; Liu, R.; Ren, Z.; Gu, S.; Lin, Z.; et al. TALENTACE: A Phase III, Open-Label, Randomized Study of on-Demand Transarterial Chemoembolization Combined with Atezolizumab + Bevacizumab or on-Demand Transarterial Chemoembolization Alone in Patients with Untreated Hepatocellular Carcinoma. J. Clin. Oncol. 2022, 40, TPS487. [Google Scholar] [CrossRef]
- Foerster, F.; Kloeckner, R.; Reig, M.; Chan, S.L.; Chung, J.W.; Merle, P.; Park, J.-W.; Piscaglia, F.; Vogel, A.; Gaillard, V.; et al. ABC-HCC: A Phase IIIb, Randomized, Multicenter, Open-Label Trial of Atezolizumab plus Bevacizumab versus Transarterial Chemoembolization (TACE) in Intermediate-Stage Hepatocellular Carcinoma. J. Clin. Oncol. 2022, 40, TPS498. [Google Scholar] [CrossRef]
- Kudo, M. Atezolizumab plus Bevacizumab Followed by Curative Conversion (ABC Conversion) in Patients with Unresectable, TACE-Unsuitable Intermediate-Stage Hepatocellular Carcinoma. Liver Cancer 2022, 11, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.A.; Patel, V.G. The Role of PD-L1 Expression as a Predictive Biomarker: An Analysis of All US Food and Drug Administration (FDA) Approvals of Immune Checkpoint Inhibitors. J. Immunother. Cancer 2019, 7, 278. [Google Scholar] [CrossRef]
- Iwata, H.; Emens, L.; Adams, S.; Barrios, C.H.; Diéras, V.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Winer, E.P.; Patel, S.; et al. 49MO IMpassion130: Final OS Analysis from the Pivotal Phase III Study of Atezolizumab + Nab-Paclitaxel vs Placebo + Nab-Paclitaxel in Previously Untreated Locally Advanced or Metastatic Triple-Negative Breast Cancer. Ann. Oncol. 2020, 31, S1261–S1262. [Google Scholar] [CrossRef]
- Zhang, X.; Ning, C.; Zhao, H. Tissue-Based PD-L1 Expression Is the Strongest Predictor of Overall Survival Benefit from ICI in Advanced Gastroesophageal Cancer. JAMA Oncol. 2023, 9, 280. [Google Scholar] [CrossRef]
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a Biomarker of Response to Immune-Checkpoint Inhibitors. Nat. Rev. Clin. Oncol. 2021, 18, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Pinato, D.J.; Mauri, F.A.; Spina, P.; Cain, O.; Siddique, A.; Goldin, R.; Victor, S.; Pizio, C.; Akarca, A.U.; Boldorini, R.L.; et al. Clinical Implications of Heterogeneity in PD-L1 Immunohistochemical Detection in Hepatocellular Carcinoma: The Blueprint-HCC Study. Br. J. Cancer 2019, 120, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Neely, J.; Sangro, B.; Finn, R.S.; Abou-Alfa, G.K.; Cheng, A.-L.; Yau, T.; Furuse, J.; Park, J.-W.; Wadhawan, S.; et al. Biomarkers and Clinical Outcomes in Nivolumab-Treated Patients with Advanced Hepatocellular Carcinoma in CheckMate 040. Ann. Oncol. 2019, 30, vi106. [Google Scholar] [CrossRef]
- Sangro, B.; Melero, I.; Wadhawan, S.; Finn, R.S.; Abou-Alfa, G.K.; Cheng, A.-L.; Yau, T.; Furuse, J.; Park, J.-W.; Boyd, Z.; et al. Association of Inflammatory Biomarkers with Clinical Outcomes in Nivolumab-Treated Patients with Advanced Hepatocellular Carcinoma. J. Hepatol. 2020, 73, 1460–1469. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib (KEYNOTE-224): A Non-Randomised, Open-Label Phase 2 Trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, D.; Zhao, B.; Ren, L.; Huang, R.; Feng, B.; Chen, H. The Predictive Value of PD-L1Expression in Patients with Advanced Hepatocellular Carcinoma Treated with PD -1/PD-L1 Inhibitors: A Systematic Review and Meta-analysis. Cancer Med. 2023, 12, 9282–9292. [Google Scholar] [CrossRef] [PubMed]
- Sha, D.; Jin, Z.; Budczies, J.; Kluck, K.; Stenzinger, A.; Sinicrope, F.A. Tumor Mutational Burden as a Predictive Biomarker in Solid Tumors. Cancer Discov. 2020, 10, 1808–1825. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Kim, J.T.; Cox, B.; Larson, B.K.; Kim, S.; Waters, K.M.; Vail, E.; Guindi, M. Evaluation of Tumor Mutational Burden in Small Early Hepatocellular Carcinoma and Progressed Hepatocellular Carcinoma. Hepat. Oncol. 2021, 8, HEP39. [Google Scholar] [CrossRef] [PubMed]
- Muhammed, A.; D’Alessio, A.; Enica, A.; Talbot, T.; Fulgenzi, C.A.M.; Nteliopoulos, G.; Goldin, R.D.; Cortellini, A.; Pinato, D.J. Predictive Biomarkers of Response to Immune Checkpoint Inhibitors in Hepatocellular Carcinoma. Expert. Rev. Mol. Diagn. 2022, 22, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Eso, Y.; Shimizu, T.; Takeda, H.; Takai, A.; Marusawa, H. Microsatellite Instability and Immune Checkpoint Inhibitors: Toward Precision Medicine against Gastrointestinal and Hepatobiliary Cancers. J. Gastroenterol. 2020, 55, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Kawaoka, T.; Ando, Y.; Yamauchi, M.; Suehiro, Y.; Yamaoka, K.; Kosaka, Y.; Fuji, Y.; Uchikawa, S.; Morio, K.; Fujino, H.; et al. Incidence of Microsatellite Instability-high Hepatocellular Carcinoma among Japanese Patients and Response to Pembrolizumab. Hepatol. Res. 2020, 50, 885–888. [Google Scholar] [CrossRef] [PubMed]
- Heimbach, J.K.; Kulik, L.M.; Finn, R.S.; Sirlin, C.B.; Abecassis, M.M.; Roberts, L.R.; Zhu, A.X.; Murad, M.H.; Marrero, J.A. AASLD Guidelines for the Treatment of Hepatocellular Carcinoma. Hepatology 2018, 67, 358–380. [Google Scholar] [CrossRef]
- Brummel, K.; Eerkens, A.L.; de Bruyn, M.; Nijman, H.W. Tumour-Infiltrating Lymphocytes: From Prognosis to Treatment Selection. Br. J. Cancer 2023, 128, 451–458. [Google Scholar] [CrossRef]
- Thomas, N.E.; Busam, K.J.; From, L.; Kricker, A.; Armstrong, B.K.; Anton-Culver, H.; Gruber, S.B.; Gallagher, R.P.; Zanetti, R.; Rosso, S.; et al. Tumor-Infiltrating Lymphocyte Grade in Primary Melanomas Is Independently Associated with Melanoma-Specific Survival in the Population-Based Genes, Environment and Melanoma Study. J. Clin. Oncol. 2013, 31, 4252–4259. [Google Scholar] [CrossRef]
- Zeng, D.-Q.; Yu, Y.-F.; Ou, Q.-Y.; Li, X.-Y.; Zhong, R.-Z.; Xie, C.-M.; Hu, Q.-G. Prognostic and Predictive Value of Tumor-Infiltrating Lymphocytes for Clinical Therapeutic Research in Patients with Non-Small Cell Lung Cancer. Oncotarget 2016, 7, 13765–13781. [Google Scholar] [CrossRef]
- Presti, D.; Dall’Olio, F.G.; Besse, B.; Ribeiro, J.M.; Di Meglio, A.; Soldato, D. Tumor Infiltrating Lymphocytes (TILs) as a Predictive Biomarker of Response to Checkpoint Blockers in Solid Tumors: A Systematic Review. Crit. Rev. Oncol. Hematol. 2022, 177, 103773. [Google Scholar] [CrossRef] [PubMed]
- Macek Jilkova, Z.; Aspord, C.; Kurma, K.; Granon, A.; Sengel, C.; Sturm, N.; Marche, P.N.; Decaens, T. Immunologic Features of Patients with Advanced Hepatocellular Carcinoma Before and During Sorafenib or Anti-Programmed Death-1/Programmed Death-L1 Treatment. Clin. Transl. Gastroenterol. 2019, 10, e00058. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.H.M.; Lee, R.Y.; Goh, S.; Tay, I.S.Y.; Lim, X.; Lee, B.; Chew, V.; Li, H.; Tan, B.; Lim, S.; et al. Immunohistochemical Scoring of CD38 in the Tumor Microenvironment Predicts Responsiveness to Anti-PD-1/PD-L1 Immunotherapy in Hepatocellular Carcinoma. J. Immunother. Cancer 2020, 8, e000987. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Cho, H.J.; Sa, J.K.; Liu, X.; Ha, S.Y.; Lee, T.; Kim, H.; Kang, W.; Sinn, D.H.; Gwak, G.-Y.; et al. Hepatocellular Carcinoma Patients with High Circulating Cytotoxic T Cells and Intra-Tumoral Immune Signature Benefit from Pembrolizumab: Results from a Single-Arm Phase 2 Trial. Genome Med. 2022, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Haber, P.K.; Castet, F.; Torres-Martin, M.; Andreu-Oller, C.; Puigvehí, M.; Miho, M.; Radu, P.; Dufour, J.-F.; Verslype, C.; Zimpel, C.; et al. Molecular Markers of Response to Anti-PD1 Therapy in Advanced Hepatocellular Carcinoma. Gastroenterology 2023, 164, 72–88.e18. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Abbas, A.R.; de Galarreta, M.R.; Guan, Y.; Lu, S.; Koeppen, H.; Zhang, W.; Hsu, C.-H.; He, A.R.; Ryoo, B.-Y.; et al. Molecular Correlates of Clinical Response and Resistance to Atezolizumab in Combination with Bevacizumab in Advanced Hepatocellular Carcinoma. Nat. Med. 2022, 28, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef]
- Haber, P.K.; Puigvehí, M.; Castet, F.; Lourdusamy, V.; Montal, R.; Tabrizian, P.; Buckstein, M.; Kim, E.; Villanueva, A.; Schwartz, M.; et al. Evidence-Based Management of Hepatocellular Carcinoma: Systematic Review and Meta-Analysis of Randomized Controlled Trials (2002–2020). Gastroenterology 2021, 161, 879–898. [Google Scholar] [CrossRef]
- Lee, C.-K.; Chan, S.L.; Chon, H.J. Could We Predict the Response of Immune Checkpoint Inhibitor Treatment in Hepatocellular Carcinoma? Cancers 2022, 14, 3213. [Google Scholar] [CrossRef]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH Limits Anti-Tumour Surveillance in Immunotherapy-Treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Kudo, M. Lack of Response to Immunotherapy in Non-Alcoholic Steatohepatitis Related Hepatocellular Carcinoma. Hepatobiliary Surg. Nutr. 2021, 10, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Chan, L.S.; Gu, S.; Bai, Y.; Ren, Z.; Lin, X.; Chen, Z.; Jia, W.; Jin, Y.; Guo, Y.; et al. LBA35 Camrelizumab (C) plus Rivoceranib (R) vs. Sorafenib (S) as First-Line Therapy for Unresectable Hepatocellular Carcinoma (UHCC): A Randomized, Phase III Trial. Ann. Oncol. 2022, 33, S1401–S1402. [Google Scholar] [CrossRef]
- Chan, S.L.; Kudo, M.; Sangro, B.; Kelley, R.K.; Furuse, J.; Park, J.-W.; Sunpaweravong, P.; Fasolo, A.; Yau, T.; Kawaoka, T.; et al. 83P Impact of Viral Aetiology in the Phase III HIMALAYA Study of Tremelimumab (T) plus Durvalumab (D) in Unresectable Hepatocellular Carcinoma (UHCC). Ann. Oncol. 2022, 33, S1465–S1466. [Google Scholar] [CrossRef]
- D’Alessio, A.; Fulgenzi, C.A.M. Treating Patients with Advanced Hepatocellular Carcinoma and Impaired Liver Function: Broadening the Reach of Anti-cancer Therapy. Liver Cancer Int. 2021, 2, 31–32. [Google Scholar] [CrossRef]
- Fessas, P.; Kaseb, A.; Wang, Y.; Saeed, A.; Szafron, D.; Jun, T.; Dharmapuri, S.; Rafeh Naqash, A.; Muzaffar, M.; Navaid, M.; et al. Post-Registration Experience of Nivolumab in Advanced Hepatocellular Carcinoma: An International Study. J. Immunother. Cancer 2020, 8, e001033. [Google Scholar] [CrossRef]
- Haanen, J.; Obeid, M.; Spain, L.; Carbonnel, F.; Wang, Y.; Robert, C.; Lyon, A.R.; Wick, W.; Kostine, M.; Peters, S.; et al. Management of Toxicities from Immunotherapy: ESMO Clinical Practice Guideline for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2022, 33, 1217–1238. [Google Scholar] [CrossRef] [PubMed]
- Pinter, M.; Scheiner, B.; Peck-Radosavljevic, M. Immunotherapy for Advanced Hepatocellular Carcinoma: A Focus on Special Subgroups. Gut 2021, 70, 204–214. [Google Scholar] [CrossRef]
- Kennedy, L.C.; Bhatia, S.; Thompson, J.A.; Grivas, P. Preexisting Autoimmune Disease: Implications for Immune Checkpoint Inhibitor Therapy in Solid Tumors. J. Natl. Compr. Cancer Netw. 2019, 17, 750–757. [Google Scholar] [CrossRef]
- Shi, X.-L.; Mancham, S.; Hansen, B.E.; de Knegt, R.J.; de Jonge, J.; van der Laan, L.J.W.; Rivadeneira, F.; Metselaar, H.J.; Kwekkeboom, J. Counter-Regulation of Rejection Activity against Human Liver Grafts by Donor PD-L1 and Recipient PD-1 Interaction. J. Hepatol. 2016, 64, 1274–1282. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Study Drug | Number of Patients | ORR (CR) % | mPFS | mOS (95% CI) | HR | Grade 3–4 TRAE (%) | Primary End Point Met? |
|---|---|---|---|---|---|---|---|
| First line | |||||||
| Checkmate 459 Yau et al., 2022 [39] | |||||||
| Nivolumab 240 mg Q2W | 371 | 15 (4) | 3.7 | 16.4 (14.0–18.5) | 0.85 | 22 | No—OS did not reach significance per specified criteria |
| Sorafenib 400 mg BD | 372 | 7 (1) | 3.8 | 14.8 (12.1–17.3) | 49 | ||
| RATIONALE 301 Qin et al., 2019 [40] | |||||||
| Tislelizumab 200 mg Q3W | 342 | 14 | 2.2 | 15.9 (13.2–19.7) | 1.1 | 11 | Yes—OS with tislelizumab non-inferior to sorafenib |
| Sorafenib 400 mg BD | 332 | 5 | 3.6 | 14.1 (12.6–17.4) | 5 | ||
| HIMALAYA Abou-Alfa et al., 2022 [41] | |||||||
| STRIDE single loading dose of 300 mg tremelimumab and durvalumab 500 mg Q4W | 393 | 20 (3) | 3.78 | 16.4 (14.1–19.58) | 0.78 (STRIDE compared to Sorafenib) | 51 | Yes—STRIDE significantly improved OS versus sorafenib. Durvalumab monotherapy was noninferior to sorafenib |
| Single-agent Durvalumab 1500 mg Q4W | 389 | 17 (2) | 3.65 | 16.6 (14.1–19.1) | 0.86 (non-inferior to Sorafenib) | 37 | |
| Sorafenib | 389 | 20 (0) | 4.07 | 13.8 (12.3–16.1) | 40 | ||
| IMBRAVE 150 Cheng et al., 2022 [42] | |||||||
| Atezolizumab 1200 mg, Q3W plus bevacizumab 5 mg/kg Q3W | 336 | 30 (8) | 6.9 | 19.2 (17.0–23.7) | 0.66 | 43 | Yes—atezolizumab combined with bevacizumab resulted in better OS and PFS than sorafenib |
| Sorafenib 400 mg BD | 165 | 11 (<1) | 4.3 | 13.4 (11.4–16.9) | 46 | ||
| ORIENT 32 Ren at al., 2021 [43] | |||||||
| Sintilimab 200 mg Q3W plus IBI305 15 mg/kg Q3W | 380 | 20 (1) | 4.6 | NE (NE-NE) | 0.57 | 34 | Yes—sintilimab plus IBI305 showed a significant OS and PFS benefit versus sorafenib |
| Sorafenib 400 mg BD | 191 | 5 (0) | 2.8 | 10.4 (8.5-NR) | 36 | ||
| COSMIC 312 Kelley et al., 2022 [44] | |||||||
| Cabozantinib 40 mg OD and atezolizumab 1200 mg Q3W | 432 | 11 (1) | 6.8 | 15.4 (13.7–17.7) | 0.90 (compared with Sorafenib) | 64 | In part—primary PFS was significantly longer in the combination treatment group versus the sorafenib group. At interim analysis OS did not differ significantly between the treatment groups |
| Cabozantinib 50 mg OD | 118 | 6 (0) | 5.8 | 46 | |||
| Sorafenib 400 mg BD | 217 | 4 (0) | 4.2 | 15.5 (12.1-NE) | 60 | ||
| CARES 310 Qin, Chan, et al., 2023 [45] | |||||||
| Camreliziumab 200 mg Q2W and rivoceranib 250 mg PO QDS | 272 | 25 (1) | 5.6 | 22.1 (19.1–27.2) | 0.62 | 81 | Yes—camrelizumab and rivoceranib significantly prolonged PFS and OS and improved ORR versus sorafenib |
| Sorafenib 400 mg BD | 271 | 6 (0.4) | 3.7 | 15.2 (13.0–18.5) | 52 | ||
| LEAP 002 Finn et al., 2022 [46] | |||||||
| Lenvatinib 8 mg or 12 mg OD plus pembrolizumab 200 mg Q3W | 395 | 26 | 8.2 | 21.2 | 0.84 | 63 | No—OS and PFS did not meet pre-specified statistical significance |
| Lenvatinib 8 mg of 12 mg OD plus placebo | 399 | 17 | 8.1 | 19.0 | 58 | ||
| Second Line | |||||||
| Keynote 240 Finn et al., 2020 [47] | |||||||
| Pembrolizumab 300 mg, Q3W | 278 | 18 (2) | 3.0 | 13.9 (11.6–16.0) | 0.78 | 53 | No—OS and PFS did not reach significance per specified criteria |
| Placebo | 135 | 4 (0) | 2.8 | 10.6 (8.3–13.5) | 46 | ||
| Keynote 394 Qin, Chen, et al., 2022 [48] | |||||||
| Pembrolizumab 200 mg Q2W | 300 | 13 | 2.9 | 14.6 (12.6–18.0) | 0.79 | 14 | Yes—pembrolizumab did significantly improve OS, PFS and ORR |
| Placebo | 153 | 1 | 2.3 | 13.0 (10.5–15.1) | 6 | ||
| Trial Name/ID | Phase | Regimen | Targets | Indication | N | Primary Endpoint | ORR | Grade 3–4 TRAE |
|---|---|---|---|---|---|---|---|---|
| NCT04444167 Bai et al., 2021 [63] | I/II | AK104 (IV 6 mg/kg Q2W) and Lenvatinib | PD-1/CTLA-4 and VEGF | First-line | 18 | ORR | ORR 44.4% DCR 77.8% | 26.7% |
| NCT03519997 Hsieh et al., 2023 [64] | II | Pembrolizumab (IV 200 mg Q3W) and Bavituximab (IV 3 mg/kg weekly) | PD-1 and anti-phosphatidylserine | First-line | 28 | ORR | ORR 32% DCR 61% | Not reported |
| RENOBATE Yoo et al., 2022 [65] | II | Nivolumab (IV 480 mg Q4W) and Regorafenib (po 80 mg daily for 21 consecutive days Q4W) | PD-1 and VEGF | First-line | 42 | ORR | ORR 31.0%. | Not reported |
| NCT03941873 Zhang et al., 2022 [66] | I | Tislelizumab (IV 200 mg Q3W) and Sitravatinib (80 mg/120 mg daily) | PD-1 and VEGF | First or later lines | 43 | Safety | ORR 10.0%. DCR 85.0% | 48.8% |
| IMMUNIB Vogel et al., 2022 [67] | II | Nivolumab (IV 240 MG Q2W up to 36 cycles) and Lenvatinib | PD-1 and VEGF | First-line | 50 | ORR | ORR 28% | 59.1% |
| GOING Sanduzzi Zamparelli et al., 2022 [68] | I/II | Nivolumab (1.5 mg/kg, 3 mg/kg or 240 mg Q2W) and Regorafenib (160 mg/day 3W on 1W off in the first 8W) | PD-1 and VEGF | Second-line | 51 | Safety | Not reported | Less than one third of the patients |
| Liver100 Kudo et al., 2021 [69] | Ib | Avelumab 10 mg/kg intravenously every 2 weeks plus Axitinib 5 mg orally twice daily | PD-1 and VEGF | First-line | 22 | Safety and ORR | ORR 13.6% | 72.7% |
| CheckMate 040 Yau et al., 2023 [70] | I/II | Nivolumab 240 mg once every 2 weeks plus Cabozantinib 40 mg once daily (doublet arm); or Nivolumab 3 mg/kg every 2 weeks plus Cabozantinib 40 mg once daily with Ipilimumab 1 mg/kg once every 6 weeks (triplet arm). | PD-1± CTLA4 and VEGF | First- or second-line | 71 | Safety ORR | ORR Doublet 17% Triplet 29% | Doublet 50% Triplet 74% |
| Study Name | Study Population (n) | Drug | Trial No/Reference |
|---|---|---|---|
| EMERALD 1 | 710 | TACE + Durvalumab + Bevacizumab vs. TACE + Durvalumab + placebo vs. TACE + placebo + placebo | NCT03778957 Sangro, Kudo, et al., 2020 [76] |
| CHECKMATE 74W | 765 | TACE + Nivolumab + Ipilimumab vs. TACE + Nivolumab + placebo vs. TACE + placebo + placebo | NCT04340193 Sangro et al., 2021 [77] |
| LEAP 012 | 950 | TACE + Pembrolizumab + Lenvatinib vs. TACE + placebo + placebo | NCT04246177 Llovet et al., 2022 [78] |
| TACE 3 | 522 | Drug eluting bead TACE + Nivolumab vs. drug-eluting bead TACE | NCT04268888 Kloeckner et al., 2021 [79] |
| TALENTACE | 342 | On-demand TACE combined with Atezolizumab + Bevacizumab vs. on-demand TACE | NCT04712643 Kudo, Guo, et al., 2022 [80] |
| ABC HCC | 434 | Atezolizumab plus Bevacizumab vs. TACE | NCT04803994 Foerster et al., 2022 [81] |
| RENOTACE | 496 | Regorafenib and Nivolumab vs. TACE | NCT04777851 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foy, V.; McNamara, M.G.; Valle, J.W.; Lamarca, A.; Edeline, J.; Hubner, R.A. Current Evidence for Immune Checkpoint Inhibition in Advanced Hepatocellular Carcinoma. Curr. Oncol. 2023, 30, 8665-8685. https://doi.org/10.3390/curroncol30090628
Foy V, McNamara MG, Valle JW, Lamarca A, Edeline J, Hubner RA. Current Evidence for Immune Checkpoint Inhibition in Advanced Hepatocellular Carcinoma. Current Oncology. 2023; 30(9):8665-8685. https://doi.org/10.3390/curroncol30090628
Chicago/Turabian StyleFoy, Victoria, Mairéad G. McNamara, Juan W. Valle, Angela Lamarca, Julien Edeline, and Richard A. Hubner. 2023. "Current Evidence for Immune Checkpoint Inhibition in Advanced Hepatocellular Carcinoma" Current Oncology 30, no. 9: 8665-8685. https://doi.org/10.3390/curroncol30090628