

A Novel Aldisine Derivative Exhibits Potential Antitumor Effects by Targeting JAK/STAT3 Signaling
Abstract
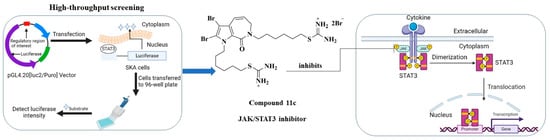
:
1. Introduction
2. Results
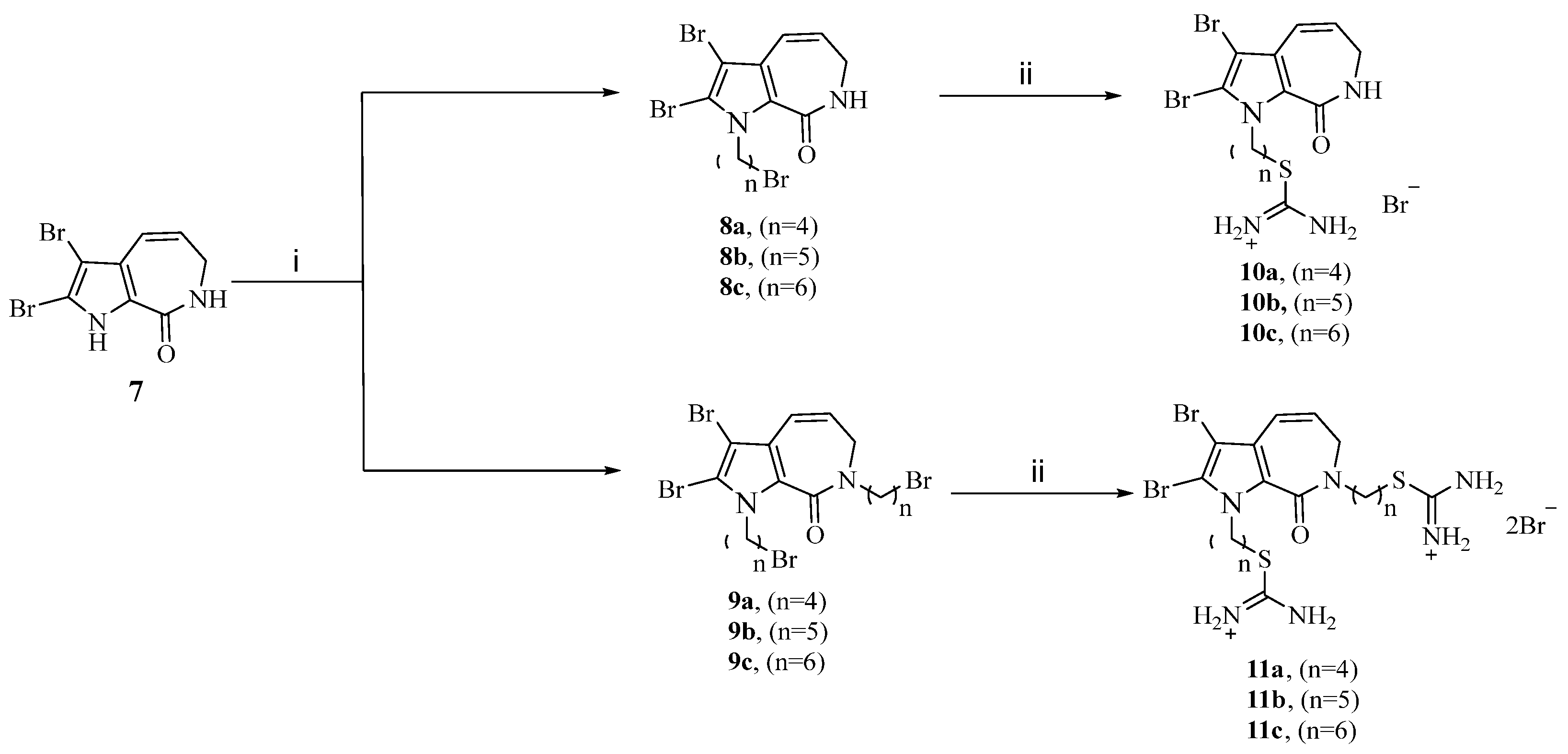
2.1. Chemistry
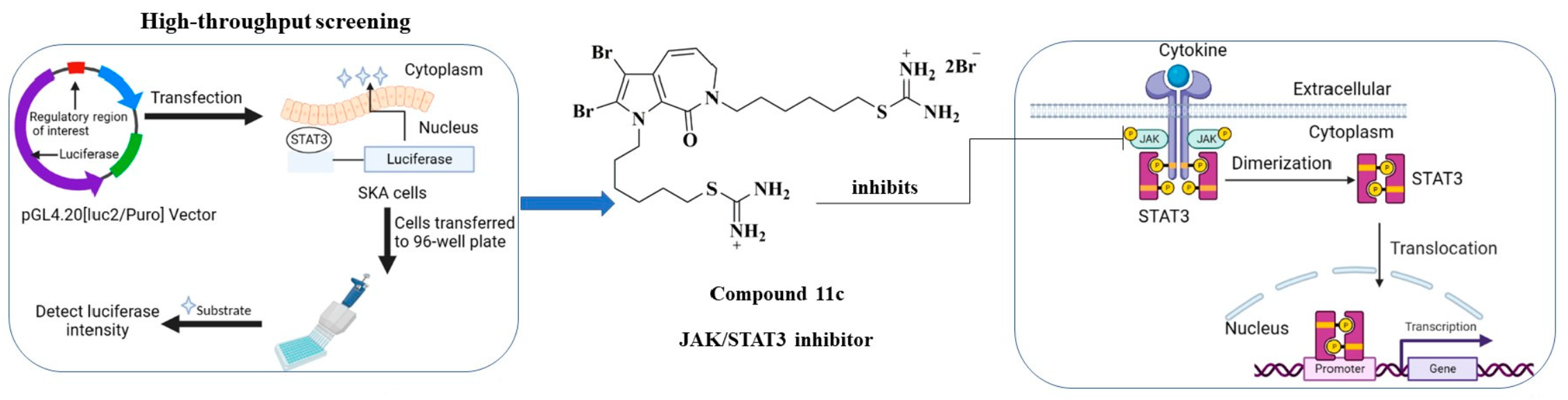
Design and Synthesis of Aldisine Derivatives
2.2. Biological Activity Assessment of Compound 11c
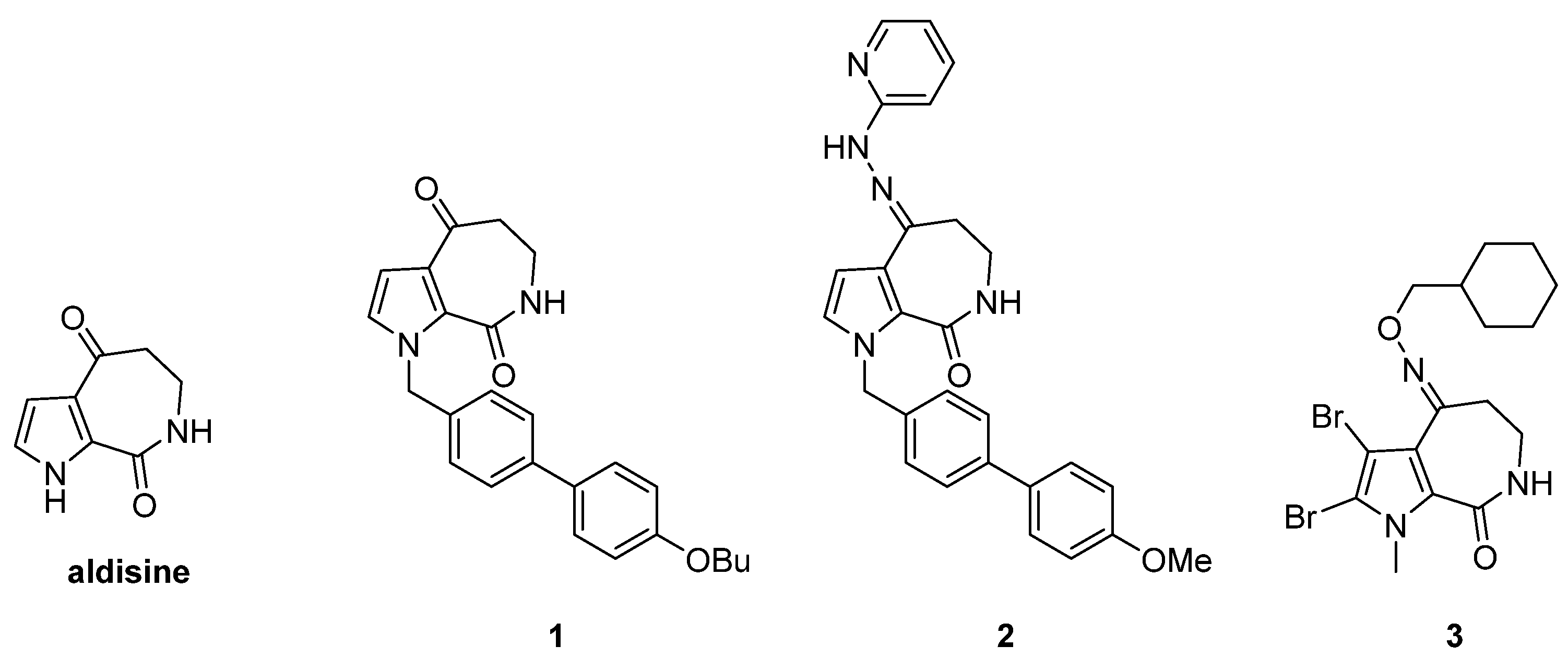
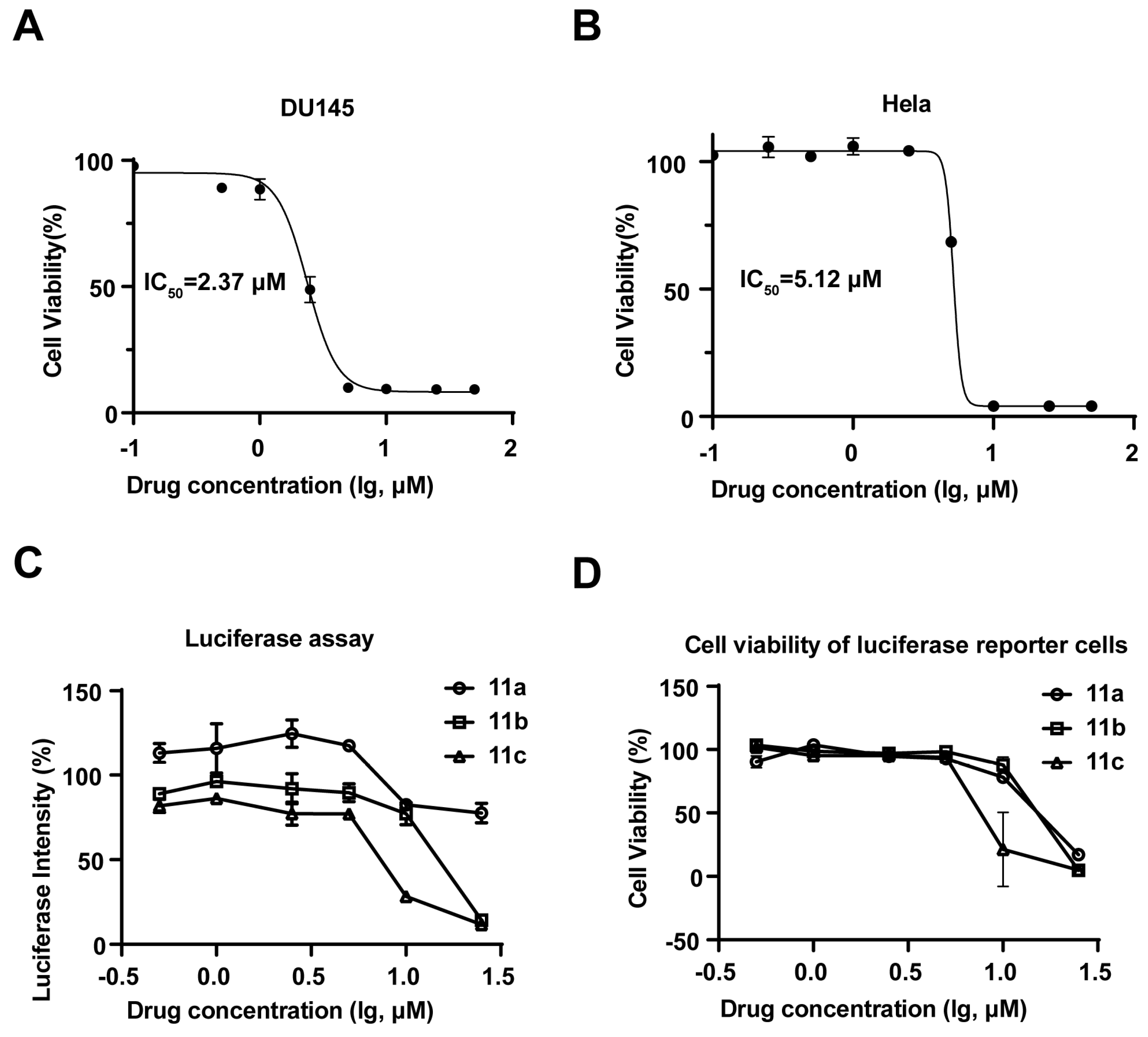
2.2.1. Compound 11c Exhibited Antiproliferative Activity and was Identified as a JAK-STAT3 Signaling Inhibitor
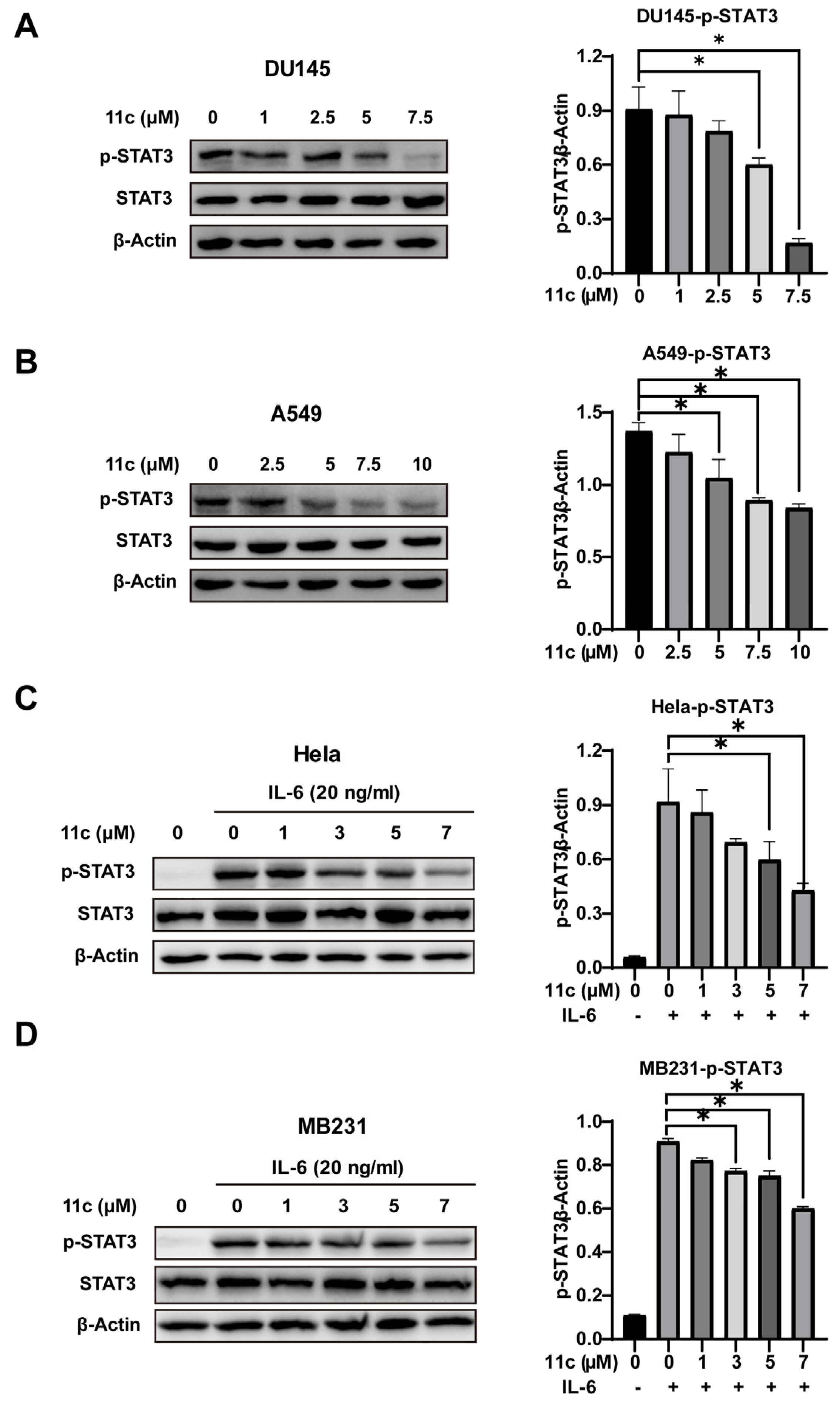
2.2.2. 11c Inhibits Constitutive and IL-6-induced STAT3 Activation
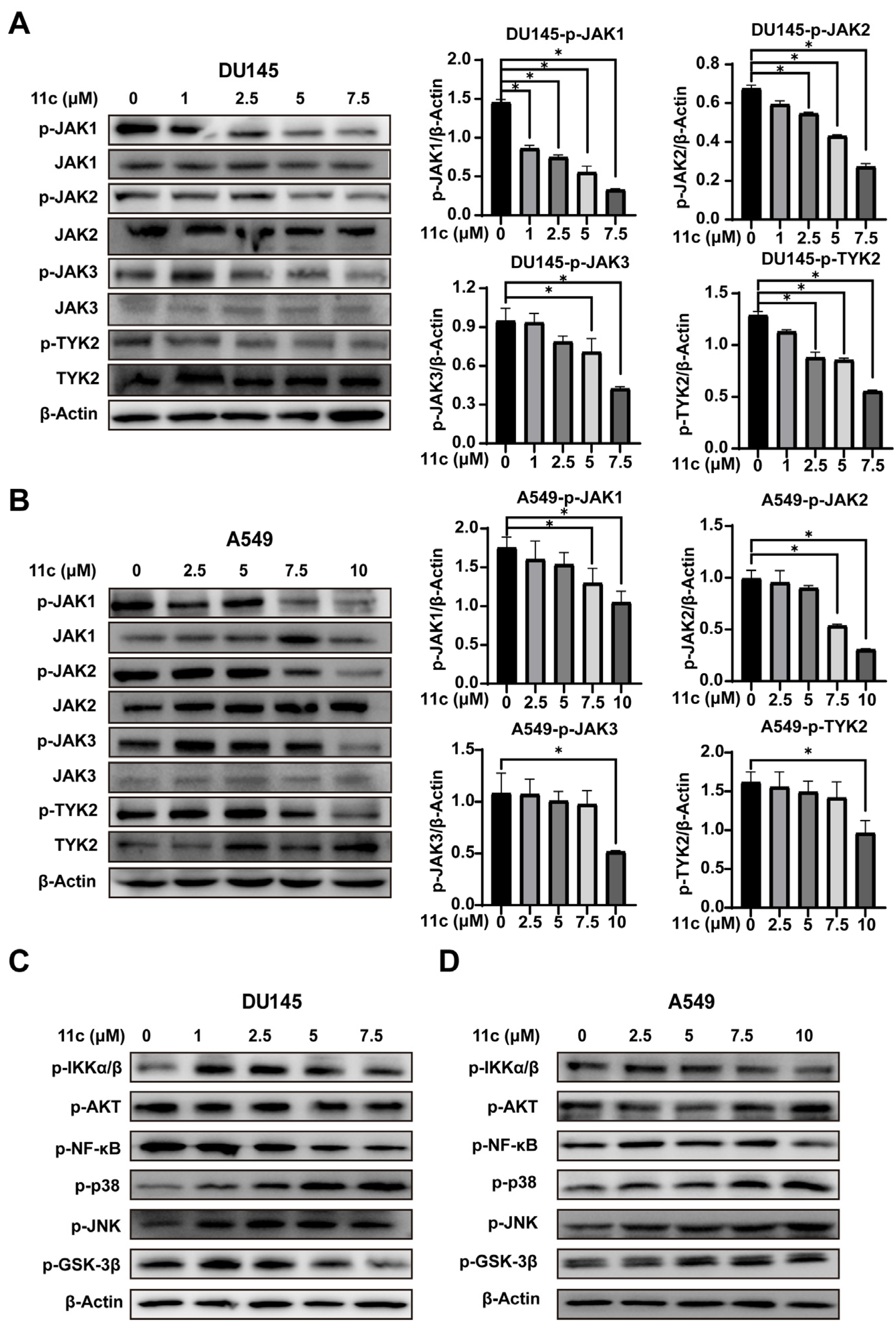
2.2.3. 11c Inhibits the Phosphorylation of JAK Family Members
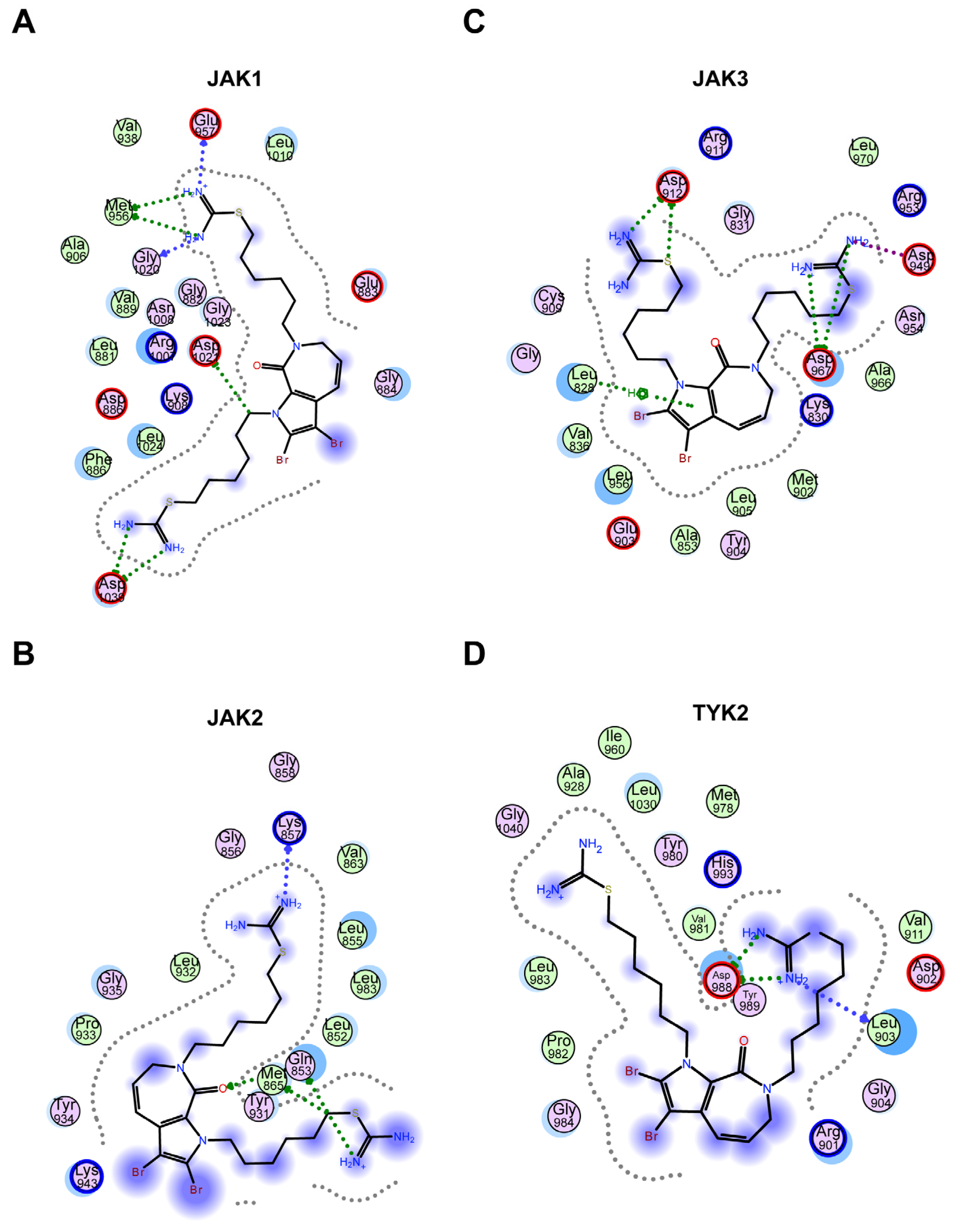
2.2.4. Molecular Docking Revealed That Hydrogen Bonding Is the Major Interaction between 11c and JAKs
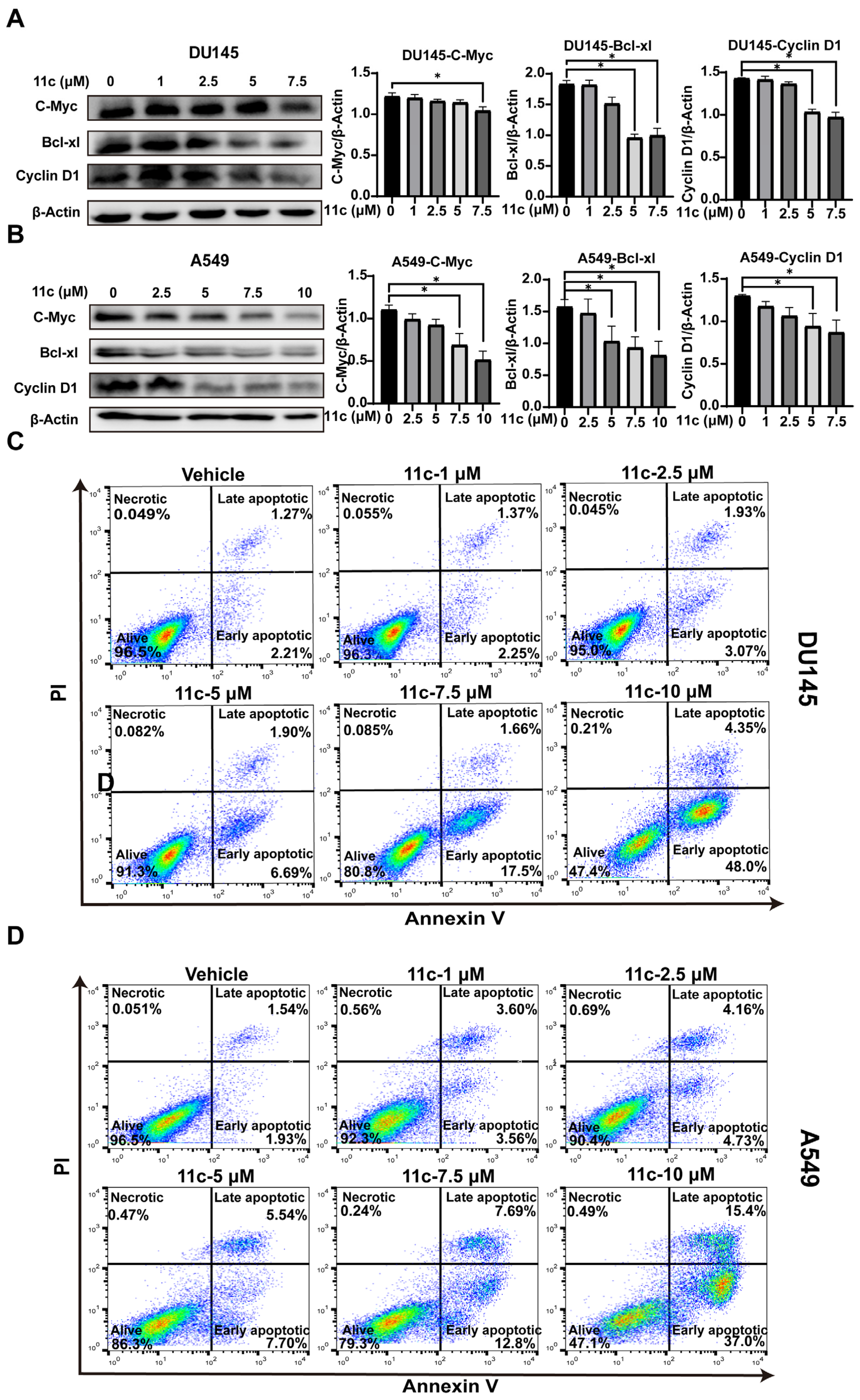
2.2.5. 11c Downregulates Anti-apoptosis Gene Expression and Induces Cancer Cell Apoptosis In Vitro
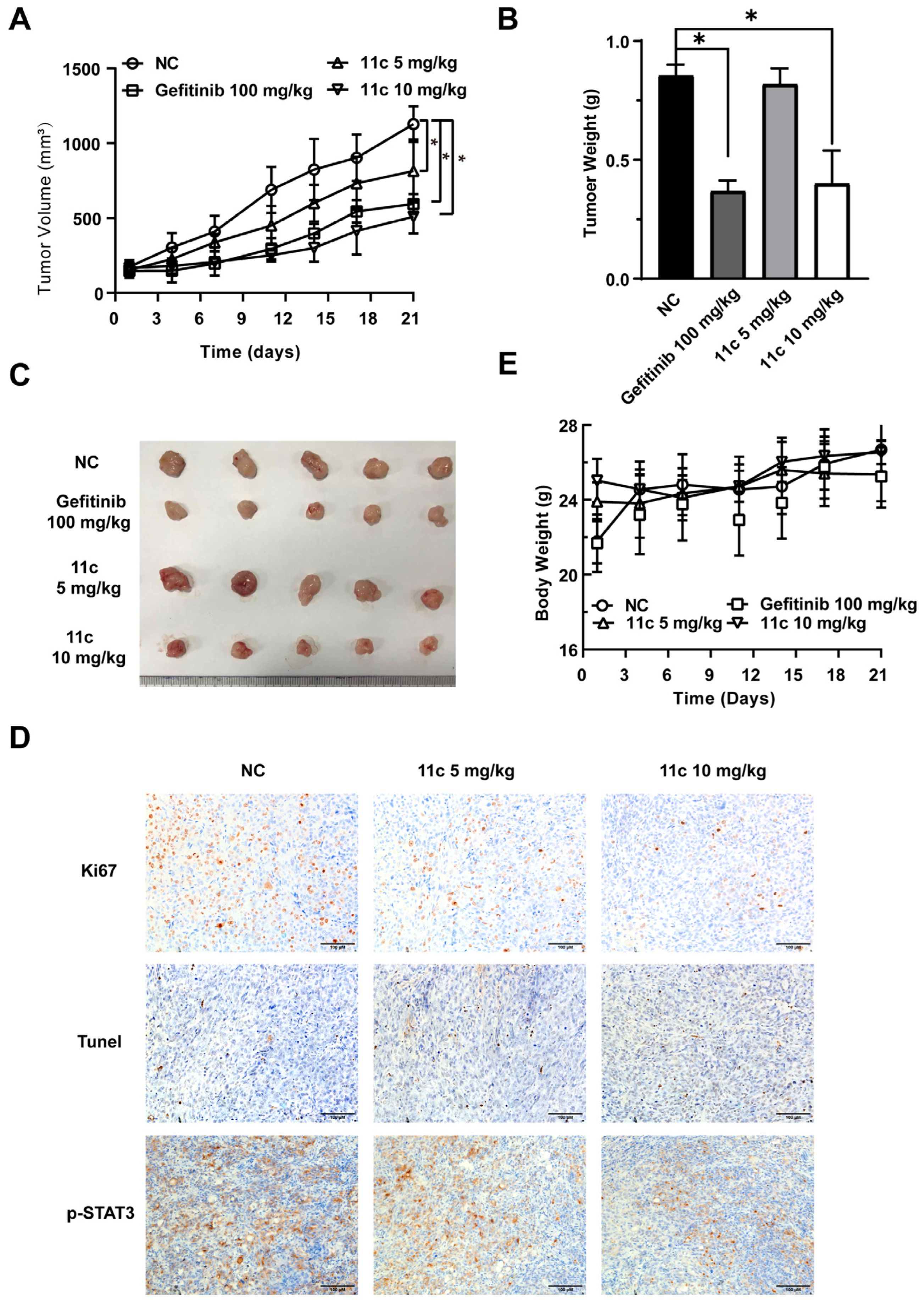
2.2.6. 11c Inhibits the Growth of DU145 Cells by Inducing Apoptosis In Vivo
3. Discussion
4. Materials and Methods
4.1. Chemical Design and Synthesis Materials
4.2. Antibodies and Reagents
4.3. Cell Culture
4.4. Animals
4.5. Luciferase Reporter Assay
4.6. Western Blot Analysis
4.7. Cell Viability and Antiproliferation Activity Assay
4.8. Molecular Docking
4.9. Flow cytometry Analysis of Apoptosis
4.10. Subcutaneous Tumor Xenograft Model and Antitumor Assay In Vivo
4.11. Immunohistochemical (IHC) Analysis
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Calautti, E.; Avalle, L.; Poli, V. Psoriasis: A STAT3-Centric View. Int. J. Mol. Sci. 2018, 19, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manore, S.G.; Doheny, D.L.; Wong, G.L.; Lo, H.-W. IL-6/JAK/STAT3 Signaling in Breast Cancer Metastasis: Biology and Treatment. Front. Oncol. 2022, 12, 866014. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, J.; Mao, Q.-F.; Wu, J.; Wang, Y. The Interaction Between Autophagy and JAK/STAT3 Signaling Pathway in Tumors. Front. Genet. 2022, 13, 880359. [Google Scholar] [CrossRef]
- Zhang, J.-Q.; Li, R.; Dong, X.-Y.; He, N.; Yin, R.-J.; Yang, M.-K.; Liu, J.-Y.; Yu, R.-L.; Zhao, C.-Y.; Jiang, T. Design, Synthesis and Structure-Activity Relationship Studies of Meridianin Derivatives as Novel JAK/STAT3 Signaling Inhibitors. Int. J. Mol. Sci. 2022, 23, 2199. [Google Scholar] [CrossRef]
- E Fragoulis, G.; McInnes, I.B.; Siebert, S. JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology 2019, 58, i43–i54. [Google Scholar] [CrossRef] [Green Version]
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef] [Green Version]
- Shawky, A.M.; Almalki, F.A.; Abdalla, A.N.; Abdelazeem, A.H.; Gouda, A.M. A Comprehensive Overview of Globally Approved JAK Inhibitors. Pharmaceutics 2022, 14, 1001. [Google Scholar] [CrossRef]
- Bryan, J.C.; Verstovsek, S. Overcoming treatment challenges in myelofibrosis and polycythemia vera: The role of ruxolitinib. Cancer Chemother. Pharmacol. 2016, 77, 1125–1142. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Yoon, D.; Song, Y.; Koh, Y.; Cao, J.; Ji, D.; Yang, H.; Eom, H.; Jing, H.; Kwak, J.; et al. Early Safety and Efficacy Data from a Phase I/II Trial of DZD4205, a Selective Jak1 Inhibitor, in Relapsed/Refractory Peripheral T-Cell Lymphoma. Hematol. Oncol. 2021, 39, 101–102. [Google Scholar] [CrossRef]
- Rigogliuso, S.; Campora, S.; Notarbartolo, M.; Ghersi, G. Recovery of Bioactive Compounds from Marine Organisms: Focus on the Future Perspectives for Pharmacological, Biomedical and Regenerative Medicine Applications of Marine Collagen. Molecules 2023, 28, 1152. [Google Scholar] [CrossRef]
- Bai, X.; Liu, Y.; Wang, H.; Zhang, H. Natural Products from the Marine Sponge Subgenus Reniera. Molecules 2021, 26, 1097. [Google Scholar] [CrossRef] [PubMed]
- Ebada, S.S.; Linh, M.H.; Longeon, A.; de Voogd, N.J.; Durieu, E.; Meijer, L.; Bourguet-Kondracki, M.L.; Singab, A.N.B.; Müller, W.E.; Proksch, P. Dispacamide E and other bioactive bromopyrrole alkaloids from two Indonesian marine sponges of the genus Stylissa. Nat. Prod. Res. 2015, 29, 231–238. [Google Scholar] [CrossRef]
- Tasdemir, D.; Mallon, R.; Greenstein, M.; Feldberg, L.R.; Kim, S.C.; Collins, K.; Wojciechowicz, D.; Mangalindan, G.C.; Concepción, G.P.; Harper, M.K.; et al. Aldisine Alkaloids from the Philippine Sponge Stylissa massa Are Potent Inhibitors of Mitogen-Activated Protein Kinase Kinase-1 (MEK-1). J. Med. Chem. 2001, 45, 529–532. [Google Scholar] [CrossRef]
- Xu, W.; Yang, R.; Hao, Y.; Song, H.; Liu, Y.; Zhang, J.; Li, Y.; Wang, Q. Discovery of Aldisine and Its Derivatives as Novel Antiviral, Larvicidal, and Antiphytopathogenic-Fungus Agents. J. Agric. Food Chem. 2022, 70, 12355–12363. [Google Scholar] [CrossRef]
- Xie, J.; Tian, J.; Su, L.; Huang, M.; Zhu, X.; Ye, F.; Wan, Y. Pyrrolo[2,3-c]azepine derivatives: A new class of potent protein tyrosine phosphatase 1B inhibitors. Bioorganic Med. Chem. Lett. 2011, 21, 4306–4309. [Google Scholar] [CrossRef]
- White, A.W.; Carpenter, N.; Lottin, J.R.; McClelland, R.A.; Nicholson, R.I. Synthesis and evaluation of novel anti-proliferative pyrroloazepinone and indoloazepinone oximes derived from the marine natural product hymenialdisine. Eur. J. Med. Chem. 2012, 56, 246–253. [Google Scholar] [CrossRef]
- Cimino, G.; De Rosa, S.; De Stefano, S.; Mazzarella, L.; Puliti, R.; Sodano, G. Isolation and X-ray crystal structure of a novel bromo-compound from two marine sponges. Tetrahedron Lett. 1982, 23, 247–259. [Google Scholar] [CrossRef]
- Wan, Y.; Hur, W.; Cho, C.Y.; Liu, Y.; Adrian, F.J.; Lozach, O.; Bach, S.; Mayer, T.; Fabbro, D.; Meijer, L.; et al. Synthesis and target identification of hymenialdisine analogs. Chem. Biol. 2004, 11, 247–259. [Google Scholar] [CrossRef]
- Bhuyan, B.K.; Scheidt, L.G.; Fraser, T.J. Cell cycle phase specificity of antitumor agents. Cancer Res 1972, 32, 398–407. [Google Scholar]
- Ferreira, M.; Assunção, L.S.; Silva, A.H.; Filippin-Monteiro, F.B.; Creczynski-Pasa, T.B.; Sá, M.M. Allylic isothiouronium salts: The discovery of a novel class of thiourea analogues with antitumor activity. Eur. J. Med. Chem. 2017, 129, 151–158. [Google Scholar] [CrossRef]
- Tong, S.; Zhang, M.; Wang, S.; Yin, R.; Yu, R.; Wan, S.; Jiang, T.; Zhang, L. Isothiouronium modification empowers pyrimidine-substituted curcumin analogs potent cytotoxicity and Golgi localization. Eur. J. Med. Chem. 2016, 123, 849–857. [Google Scholar] [CrossRef]
- Xu, Y.Z.; Yakushijin, K.; Horne, D.A. Synthesis of C11N5 marine sponge alkaloids:(±)-Hymenin, stevensine, hymenialdisine, and debromohymenialdisine. J. Org. Chem. 1997, 62, 456–464. [Google Scholar] [CrossRef]
- Suter, M.A.; Tan, N.Y.; Thiam, C.H.; Khatoo, M.; MacAry, P.A.; Angeli, V.; Gasser, S.; Zhang, Y.L. cGAS-STING cytosolic DNA sensing pathway is suppressed by JAK2-STAT3 in tumor cells. Sci. Rep. 2021, 11, 7243. [Google Scholar] [CrossRef]
- Yeh, H.-H.; Lai, W.-W.; Chen, H.H.W.; Liu, H.-S.; Su, W.-C. Autocrine IL-6-induced Stat3 activation contributes to the pathogenesis of lung adenocarcinoma and malignant pleural effusion. Oncogene 2006, 25, 4300–4309. [Google Scholar] [CrossRef] [Green Version]
- Schabath, M.B.; Cote, M.L. Cancer Progress and Priorities: Lung Cancer. Cancer Epidemiol. Biomark. Prev. A Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2019, 28, 1563–1579. [Google Scholar] [CrossRef] [Green Version]
- Nagpal, J.K.; Mishra, R.; Das, B.R. Activation of Stat-3 as one of the early events in tobacco chewing-mediated oral carcinogene-sis. Cancer 2002, 94, 2393–2400. [Google Scholar] [CrossRef]
- Suradej, B.; Sookkhee, S.; Panyakaew, J.; Mungkornasawakul, P.; Wikan, N.; Smith, D.R.; Potikanond, S.; Nimlamool, W. Kaempferia parviflora Extract Inhibits STAT3 Activation and Interleukin-6 Production in HeLa Cervical Cancer Cells. Int. J. Mol. Sci. 2019, 20, 4226. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Song, Q.; Zhang, X.; Li, L.; Xu, X.; Xu, X.; Li, X.; Wang, Z.; Lin, Y.; Li, X.; et al. Zelnorm, an agonist of 5-Hydroxytryptamine 4-receptor, acts as a potential antitumor drug by targeting JAK/STAT3 signaling. Investig. New Drugs 2019, 38, 311–320. [Google Scholar] [CrossRef]
- Aaronson, D.S.; Horvath, C.M. A Road Map for Those Who Don’t Know JAK-STAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef]
- Pellegrini, S.; Dusanter-Fourt, I. The Structure, Regulation and Function of the Janus Kinases (JAKs) and the Signal Transducers and Activators of Transcription (STATs). JBIC J. Biol. Inorg. Chem. 1997, 248, 615–633. [Google Scholar] [CrossRef]
- Glassman, C.R.; Tsutsumi, N.; Saxton, R.A.; Lupardus, P.J.; Jude, K.M.; Garcia, K.C. Structure of a Janus kinase cytokine receptor complex reveals the basis for dimeric activation. Science 2022, 376, 163–169. [Google Scholar] [CrossRef]
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 2020, 80, 106210. [Google Scholar] [CrossRef]
- de Araujo, E.D.; Orlova, A.; Neubauer, H.A.; Bajusz, D.; Seo, H.-S.; Dhe-Paganon, S.; Keserű, G.M.; Moriggl, R.; Gunning, P.T. Structural Implications of STAT3 and STAT5 SH2 Domain Mutations. Cancers 2019, 11, 1757. [Google Scholar] [CrossRef] [Green Version]
- Fukada, T.; Ohtani, T.; Yoshida, Y.; Shirogane, T.; Nishida, K.; Nakajima, K.; Hibi, M.; Hirano, T. STAT3 orchestrates contradictory signals in cytokine-induced G1 to S cell-cycle transition. EMBO J. 1998, 17, 6670–6677. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.P.; Chan, C.M.; Tung, L.N.; Wang, H.K.; Law, S. Tumor xenograft animal models for esophageal squamous cell carcinoma. J. Biomed. Sci. 2018, 25, 66. [Google Scholar] [CrossRef]
- Bürtin, F.; Mullins, C.S.; Linnebacher, M. Mouse models of colorectal cancer: Past, present and future perspectives. World J. Gastroenterol. 2020, 26, 1394–1426. [Google Scholar] [CrossRef]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef]
- McLornan, D.P.; E Pope, J.; Gotlib, J.; Harrison, C.N. Current and future status of JAK inhibitors. Lancet 2021, 398, 803–816. [Google Scholar] [CrossRef]
- Taylor, P.C.; Keystone, E.C.; Van Der Heijde, D.; Weinblatt, M.E.; Del Carmen Morales, L.; Gonzaga, J.R.; Yakushin, S.; Ishii, T.; Emoto, K.; Beattie, S.; et al. Baricitinib versus Placebo or Adalimumab in Rheumatoid Arthritis. N. Engl. J. Med. 2017, 376, 652–662. [Google Scholar] [CrossRef]
- Chovatiya, R.; Paller, A.S. JAK inhibitors in the treatment of atopic dermatitis. J. Allergy Clin. Immunol. 2021, 148, 927–940. [Google Scholar] [CrossRef]
- Keenan, C.; Nichols, K.E.; Albeituni, S. Use of the JAK Inhibitor Ruxolitinib in the Treatment of Hemophagocytic Lymphohis-tiocytosis. Front. Immunol. 2021, 12, 614704. [Google Scholar] [CrossRef]
- Clarke, B.; Yates, M.; Adas, M.; Bechman, K.; Galloway, J. The safety of JAK-1 inhibitors. Rheumatology 2021, 60, ii24–ii30. [Google Scholar] [CrossRef]
- Traves, P.G.; Murray, B.; Campigotto, F.; Galien, R.; Meng, A.; A Di Paolo, J. JAK selectivity and the implications for clinical inhibition of pharmacodynamic cytokine signalling by filgotinib, upadacitinib, tofacitinib and baricitinib. Ann. Rheum. Dis. 2021, 80, 865–875. [Google Scholar] [CrossRef]
- Virtanen, A.T.; Haikarainen, T.; Raivola, J.; Silvennoinen, O. Selective JAKinibs: Prospects in Inflammatory and Autoimmune Diseases. BioDrugs Clin. Immuno-Ther. Biopharm. Gene Ther. 2019, 33, 15–32. [Google Scholar] [CrossRef] [Green Version]
- Smyth, L.A.; Collins, I. Measuring and interpreting the selectivity of protein kinase inhibitors. J. Chem. Biol. 2009, 2, 131–151. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Du, Y.; Nan, J.; Zhang, X.; Qin, X.; Wang, Y.; Hou, J.; Wang, Q.; Yang, J. Brevilin A, a Novel Natural Product, Inhibits Janus Kinase Activity and Blocks STAT3 Signaling in Cancer Cells. PLoS ONE 2013, 8, e63697. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Yan, Y.; Zhang, X.; Zhang, Y.; Xu, X.; Zhang, L.; Lu, L.; Wang, J.; Zhang, Y.; Song, Q.; et al. Scaffold compound L971 exhibits anti-inflammatory activities through inhibition of JAK/STAT and NFκB signalling pathways. J. Cell. Mol. Med. 2021, 25, 6333–6347. [Google Scholar] [CrossRef]
- Kumar, P.; Nagarajan, A.; Uchil, P. Analysis of Cell Viability by the alamarBlue Assay. Cold Spring Harb. Protoc. 2018, 2018, pdb.prot095489. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.C.C.; Nerurkar, S.N.; Cai, H.Y.; Ng, H.H.M.; Wu, D.; Wee, Y.T.F.; Lim, J.C.T.; Yeong, J.; Lim, T.K.H. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun. 2020, 40, 135–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 value (μM) 1 | |||
|---|---|---|---|---|
| DU145 | A549 | Hela | MDA-MB231 | |
| 11a | 29.09 ± 4.21 | 8.48 ± 0.31 | 8.10 ± 1.27 | 12.59 ± 0.17 |
| 11b | 7.28 ± 0.13 | 7.60 ± 0.08 | 12.14 ± 0.01 | 4.46 ± 0.22 |
| 11c | 2.37 ± 0.09 | 3.62 ± 0.00 | 5.12 ± 0.37 | 3.49 ± 0.09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.-P.; Wu, L.-H.; Li, R.; He, N.; Zhang, Q.-Y.; Zhao, C.-Y.; Jiang, T. A Novel Aldisine Derivative Exhibits Potential Antitumor Effects by Targeting JAK/STAT3 Signaling. Mar. Drugs 2023, 21, 218. https://doi.org/10.3390/md21040218
Wang D-P, Wu L-H, Li R, He N, Zhang Q-Y, Zhao C-Y, Jiang T. A Novel Aldisine Derivative Exhibits Potential Antitumor Effects by Targeting JAK/STAT3 Signaling. Marine Drugs. 2023; 21(4):218. https://doi.org/10.3390/md21040218
Chicago/Turabian StyleWang, Dong-Ping, Li-Hong Wu, Rui Li, Na He, Qian-Yue Zhang, Chen-Yang Zhao, and Tao Jiang. 2023. "A Novel Aldisine Derivative Exhibits Potential Antitumor Effects by Targeting JAK/STAT3 Signaling" Marine Drugs 21, no. 4: 218. https://doi.org/10.3390/md21040218