

Characterization of the Native Disulfide Isomers of the Novel χ-Conotoxin PnID: Implications for Further Increasing Conotoxin Diversity
 , ,
, ,  ,
,
Abstract
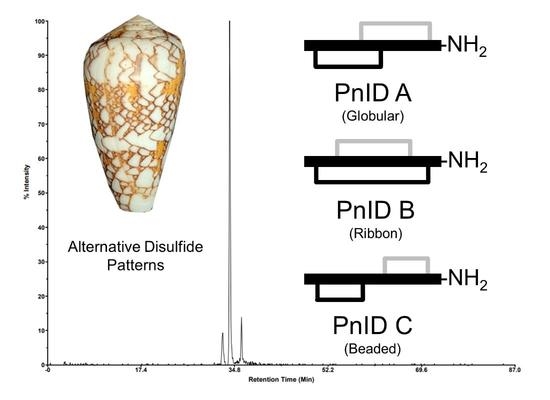
:
1. Introduction
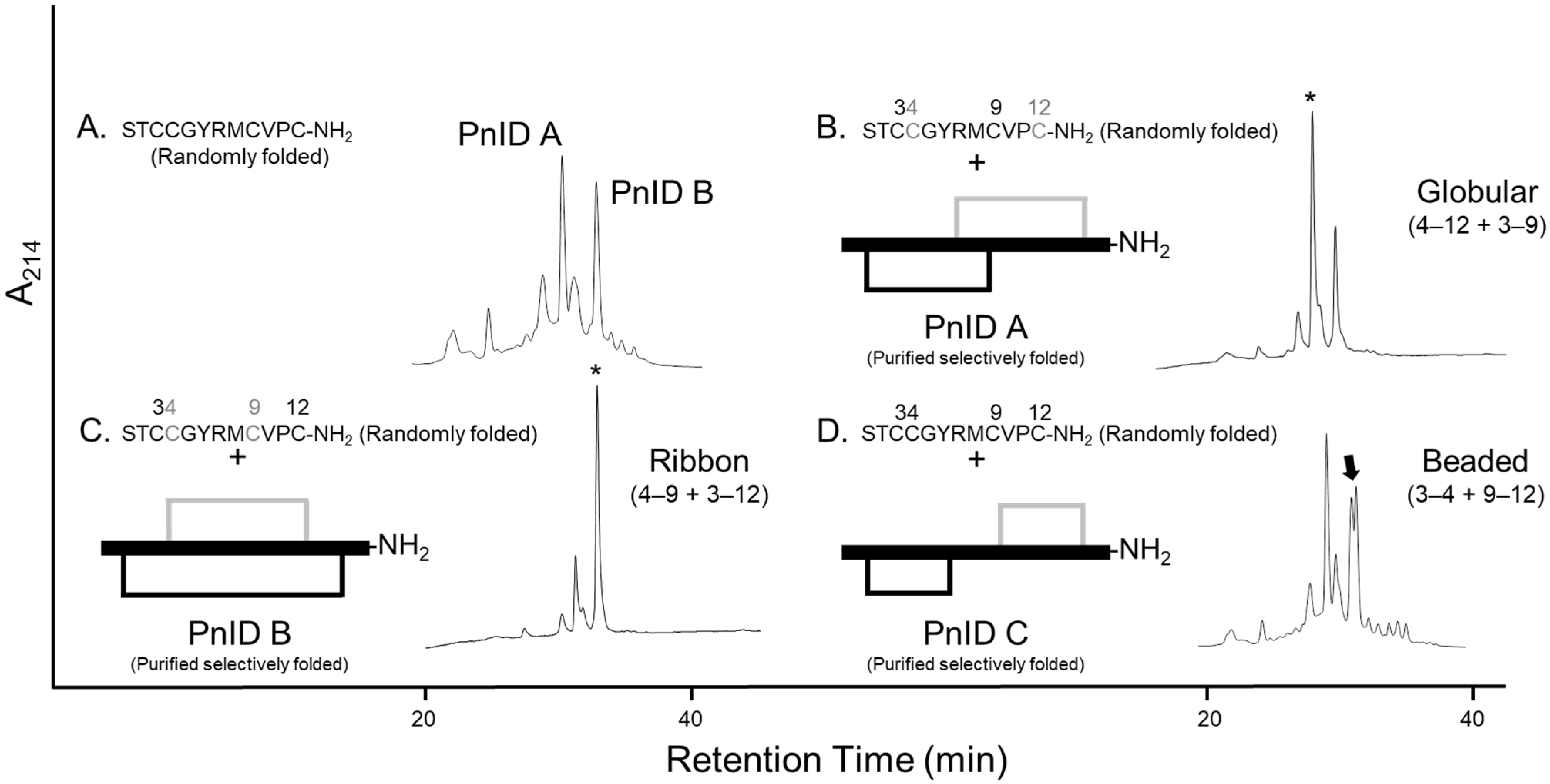
2. Results
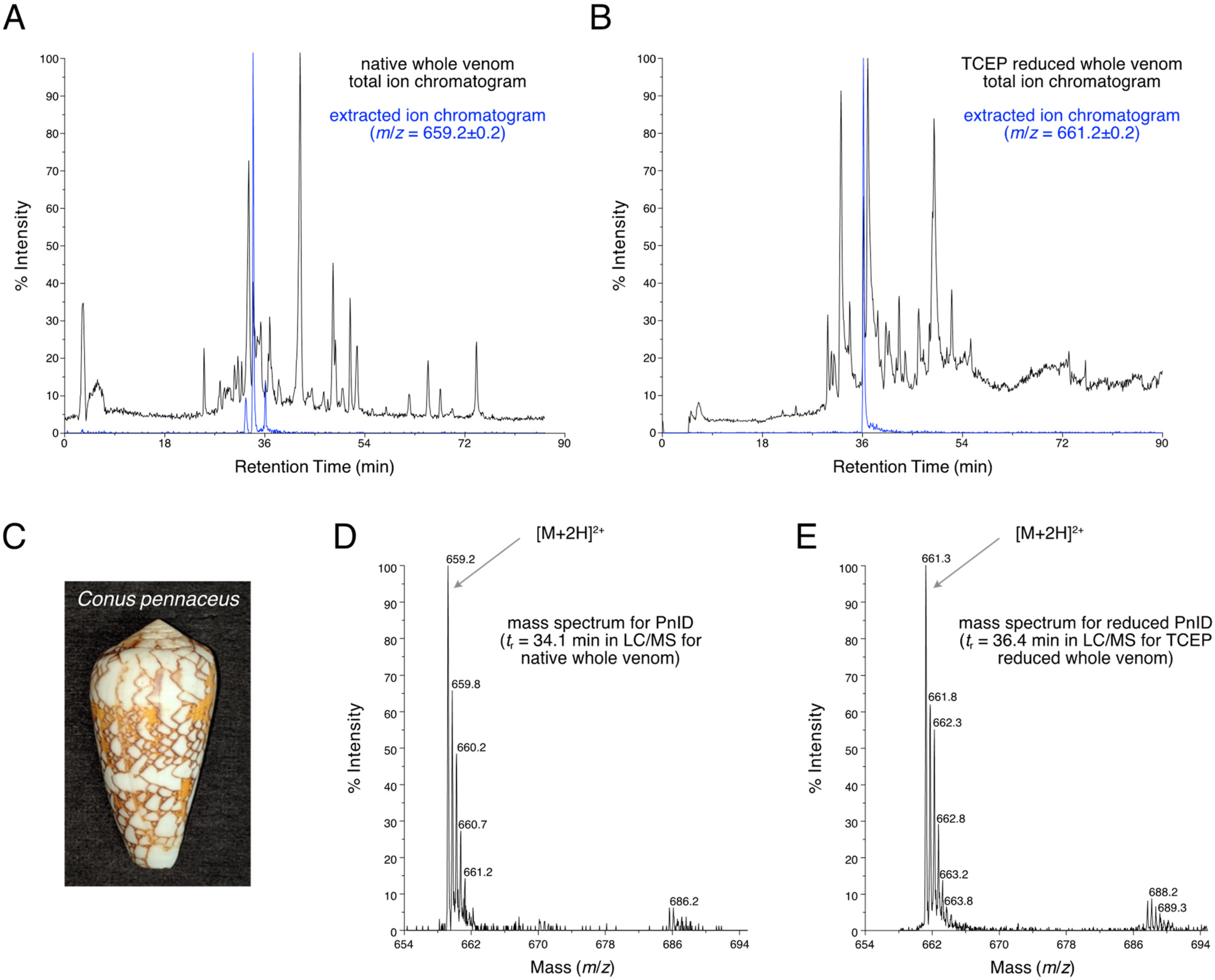
2.1. Isolation and Identification of Native Isomers
2.2. Disulfide Bond Determination by HPLC Co-Elution Experiments
2.3. Disulfide Bond Determination by Reduction and Alkylation
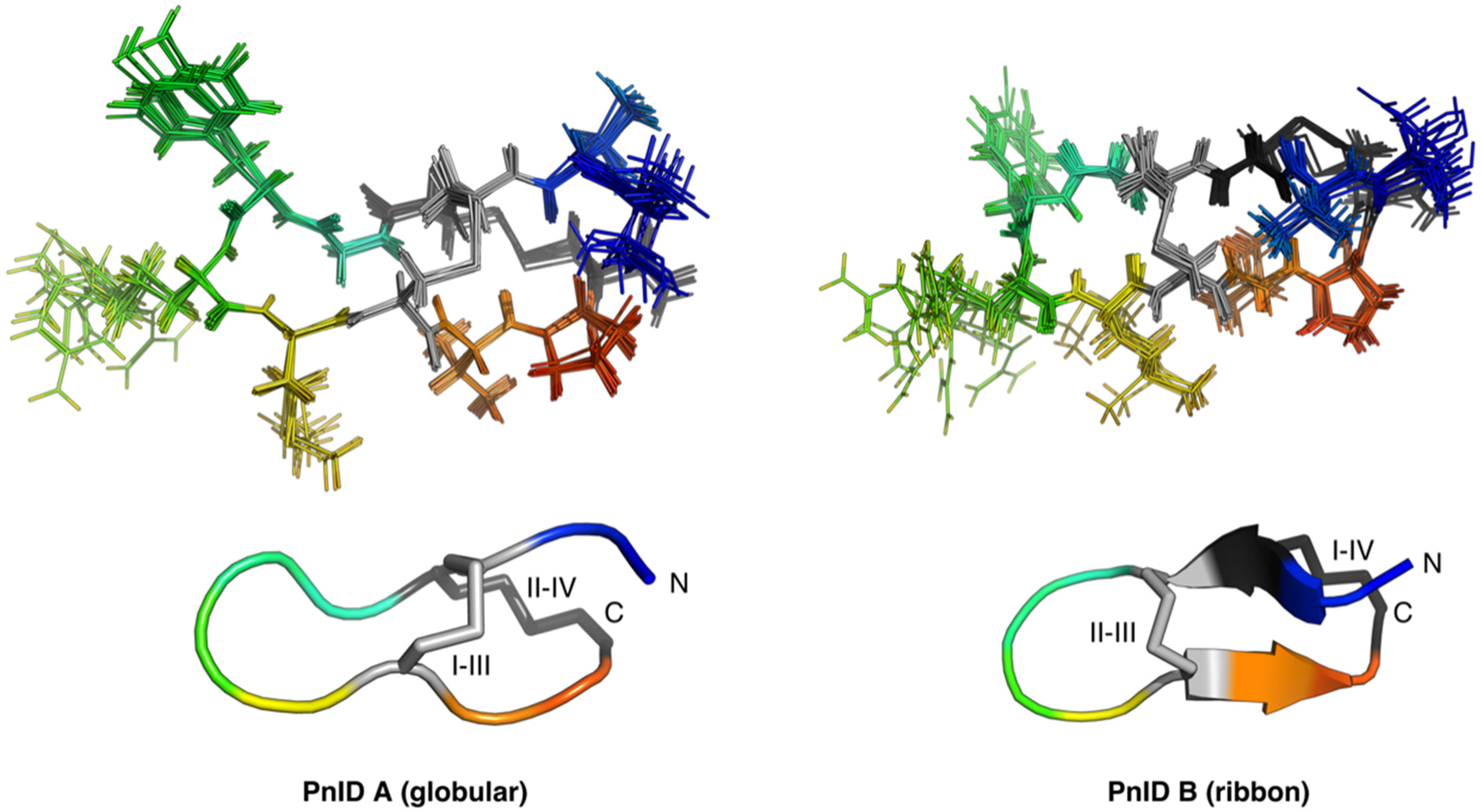
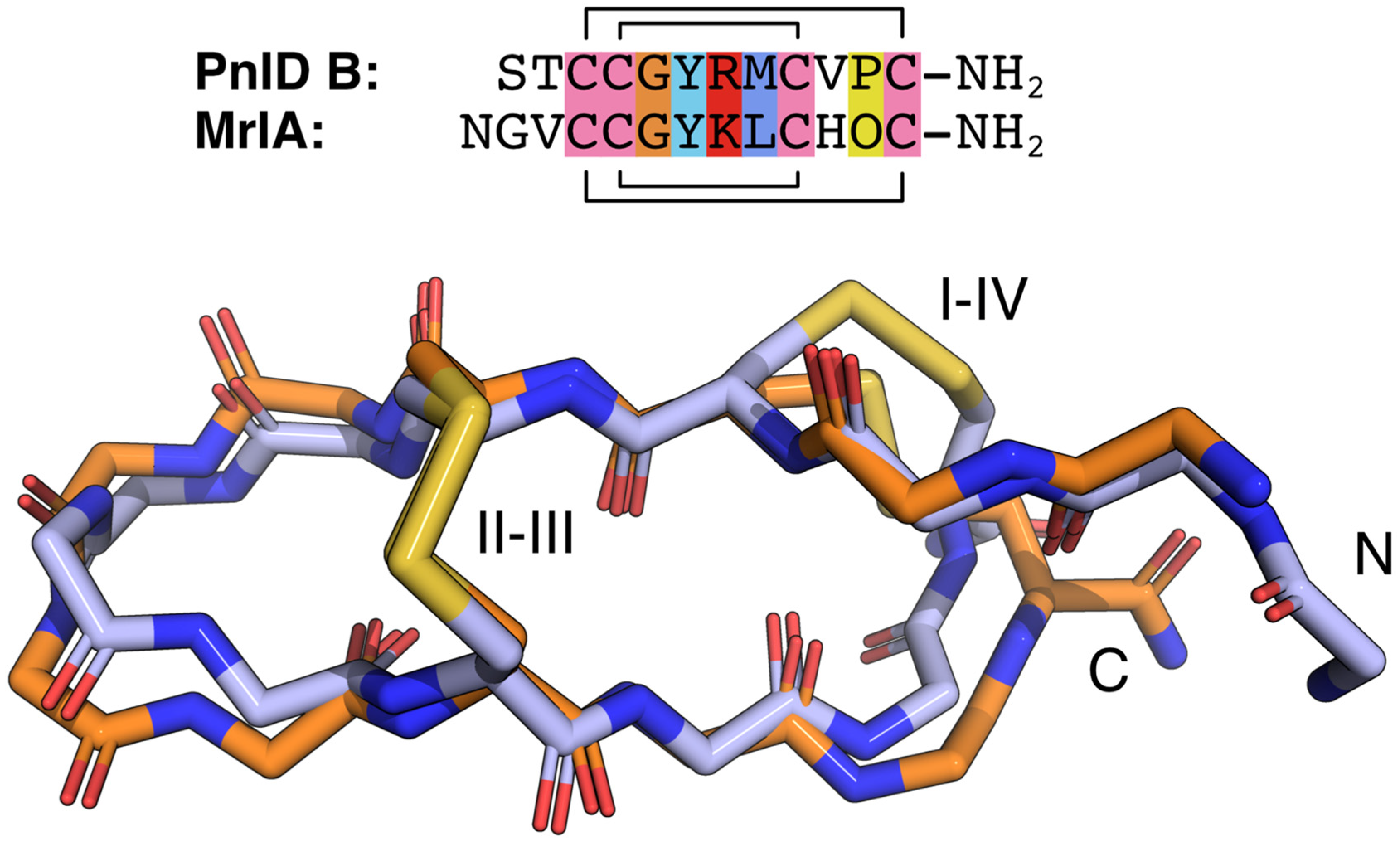
2.4. Three-Dimensional Structures of PnID
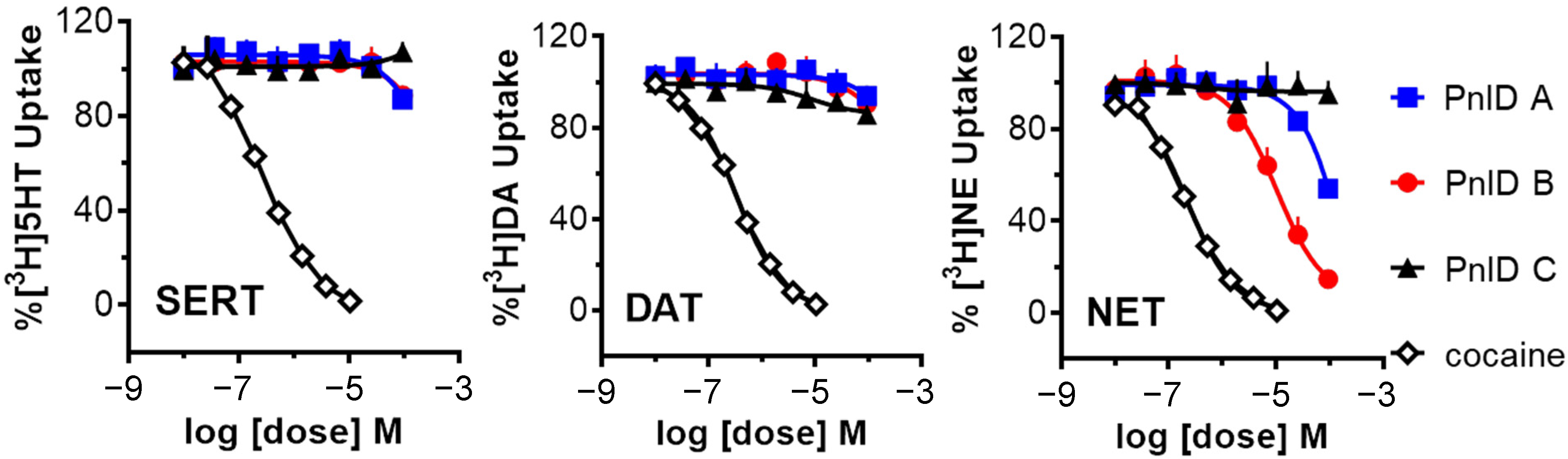
2.5. Pharmacology of the PnID Isomers
3. Discussion
4. Materials and Methods
4.1. Venom Duct Extraction
4.2. Chromatographic Separation
4.3. Disulfide Bond Connectivity Analysis by Reduction and Alkylation
4.4. Synthetic Peptide Production
4.5. Disulfide Bond Connectivity Analysis by HPLC Co-Elution
4.6. Animal Care
4.7. Pharmacology
4.8. NMR Measurements and Analysis
4.9. Structure Calculation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaas, Q.; Westermann, J.-C.; Halai, R.; Wang, C.K.L.; Craik, D.J. ConoServer, a Database for Conopeptide Sequences and Structures. Bioinformatics 2008, 24, 445–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaas, Q.; Yu, R.; Jin, A.-H.; Dutertre, S.; Craik, D.J. ConoServer: Updated Content, Knowledge, and Discovery Tools in the Conopeptide Database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.-H.; Muttenthaler, M.; Dutertre, S.; Himaya, S.W.A.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Conotoxins: Chemistry and Biology. Chem. Rev. 2019, 119, 11510–11549. [Google Scholar] [CrossRef]
- Lavergne, V.; Harliwong, I.; Jones, A.; Miller, D.; Taft, R.J.; Alewood, P.F. Optimized Deep-Targeted Proteotranscriptomic Profiling Reveals Unexplored Conus Toxin Diversity and Novel Cysteine Frameworks. Proc. Natl. Acad. Sci. USA 2015, 112, E3782–E3791. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J. Norepinephrine Transporter Inhibitors and Their Therapeutic Potential. Drugs Future 2004, 29, 1235–1244. [Google Scholar] [CrossRef]
- Balaji, R.A.; Ohtake, A.; Sato, K.; Gopalakrishnakone, P.; Kini, R.M.; Seow, K.T.; Bay, B.H. Lambda-Conotoxins, a New Family of Conotoxins with Unique Disulfide Pattern and Protein Folding. Isolation and Characterization from the Venom of Conus marmoreus. J. Biol. Chem. 2000, 275, 39516–39522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpe, I.A.; Gehrmann, J.; Loughnan, M.L.; Thomas, L.; Adams, D.A.; Atkins, A.; Palant, E.; Craik, D.J.; Adams, D.J.; Alewood, P.F.; et al. Two New Classes of Conopeptides Inhibit the Alpha1-Adrenoceptor and Noradrenaline Transporter. Nat. Neurosci. 2001, 4, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Conticello, S.G.; Gilad, Y.; Avidan, N.; Ben-Asher, E.; Levy, Z.; Fainzilber, M. Mechanisms for Evolving Hypervariability: The Case of Conopeptides. Mol. Biol. Evol. 2001, 18, 120–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, K.P.R.; Lovelace, E.S.; Caesar, C.E.; Tynngård, N.; Alewood, P.F.; Johansson, H.M.; Sharpe, I.A.; Lewis, R.J.; Daly, N.L.; Craik, D.J. Solution Structure of Chi-Conopeptide MrIA, a Modulator of the Human Norepinephrine Transporter. Biopolymers 2005, 80, 815–823. [Google Scholar] [CrossRef]
- Brust, A.; Palant, E.; Croker, D.E.; Colless, B.; Drinkwater, R.; Patterson, B.; Schroeder, C.I.; Wilson, D.; Nielsen, C.K.; Smith, M.T.; et al. χ-Conopeptide Pharmacophore Development: Toward a Novel Class of Norepinephrine Transporter Inhibitor (Xen2174) for Pain. J. Med. Chem. 2009, 52, 6991–7002. [Google Scholar] [CrossRef]
- Richardson, J.S.; Videau, L.L.; Williams, C.J.; Richardson, D.C. Broad Analysis of Vicinal Disulfides: Occurrences, Conformations with Cis or with Trans Peptides, and Functional Roles Including Sugar Binding. J. Mol. Biol. 2017, 429, 1321–1335. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.S.; Radić, Z.; Talley, T.T.; Jois, S.D.S.; Taylor, P.; Kini, R.M. Protein Folding Determinants: Structural Features Determining Alternative Disulfide Pairing in Alpha- and Chi/Lambda-Conotoxins. Biochemistry 2007, 46, 3338–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, A.-H.; Brandstaetter, H.; Nevin, S.T.; Tan, C.C.; Clark, R.J.; Adams, D.J.; Alewood, P.F.; Craik, D.J.; Daly, N.L. Structure of Alpha-Conotoxin BuIA: Influences of Disulfide Connectivity on Structural Dynamics. BMC Struct. Biol. 2007, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Dutton, J.L.; Bansal, P.S.; Hogg, R.C.; Adams, D.J.; Alewood, P.F.; Craik, D.J. A New Level of Conotoxin Diversity, a Non-Native Disulfide Bond Connectivity in Alpha-Conotoxin AuIB Reduces Structural Definition but Increases Biological Activity. J. Biol. Chem. 2002, 277, 48849–48857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoo, K.K.; Gupta, K.; Green, B.R.; Zhang, M.-M.; Watkins, M.; Olivera, B.M.; Balaram, P.; Yoshikami, D.; Bulaj, G.; Norton, R.S. Distinct Disulfide Isomers of μ-Conotoxins KIIIA and KIIIB Block Voltage-Gated Sodium Channels. Biochemistry 2012, 51, 9826–9835. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.-Y.; Speicher, D.W. Determination of Disulfide-Bond Linkages in Proteins. Curr. Protoc. Protein Sci. 2004, 37, 11.11.1–11.11.20. [Google Scholar] [CrossRef]
- Armishaw, C.J.; Singh, N.; Medina-Franco, J.L.; Clark, R.J.; Scott, K.C.M.; Houghten, R.A.; Jensen, A.A. A Synthetic Combinatorial Strategy for Developing α-Conotoxin Analogs as Potent A7 Nicotinic Acetylcholine Receptor Antagonists *. J. Biol. Chem. 2010, 285, 1809–1821. [Google Scholar] [CrossRef] [Green Version]
- Ziegman, R.; Brust, A.; Jha, P.; Cardoso, F.C.; Lewis, R.J.; Alewood, P.F. “Messy” Processing of χ-Conotoxin MrIA Generates Homologues with Reduced HNET Potency. Mar. Drugs 2019, 17, 165. [Google Scholar] [CrossRef] [Green Version]
- Okkerse, P.; Hay, J.L.; Sitsen, E.; Dahan, A.; Klaassen, E.; Houghton, W.; Groeneveld, G.J. Pharmacokinetics and Pharmacodynamics of Intrathecally Administered Xen2174, a Synthetic Conopeptide with Norepinephrine Reuptake Inhibitor and Analgesic Properties. Br. J. Clin. Pharmacol. 2017, 83, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Chun, J.B.S.; Baker, M.R.; Kim, D.H.; Leroy, M.; Toribo, P.; Bingham, J.-P. Cone Snail Milked Venom Dynamics—A Quantitative Study of Conus purpurascens. Toxicon 2012, 60, 83–94. [Google Scholar] [CrossRef]
- Bingham, J.-P.; Broxton, N.M.; Livett, B.G.; Down, J.G.; Jones, A.; Moczydlowski, E.G. Optimizing the Connectivity in Disulfide-Rich Peptides: Alpha-Conotoxin SII as a Case Study. Anal. Biochem. 2005, 338, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Atherton, D.; Fernandez, J.; DeMott, M.; Andrews, L.; Mische, S.M. ROUTINE PROTEIN SEQUENCE ANALYSIS BELOW TEN PICOMOLES: ONE SEQUENCING FACILITY’S APPROACH. In Techniques in Protein Chemistry IV; Angeletti, R.H., Ed.; Academic Press: Cambridge, MA, USA, 1993; pp. 409–418. ISBN 978-0-12-058757-5. [Google Scholar]
- Matsudaira, P.; Matsudaira, P.T. A Practical Guide to Protein and Peptide Purification for Microsequencing; Gulf Professional Publishing: Houston, TX, USA, 1993; ISBN 0-12-480282-6. [Google Scholar]
- Kapono, C.A.; Thapa, P.; Cabalteja, C.C.; Guendisch, D.; Collier, A.C.; Bingham, J.-P. Conotoxin Truncation as a Post-Translational Modification to Increase the Pharmacological Diversity within the Milked Venom of Conus magus. Toxicon 2013, 70, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.H.; Partilla, J.S.; Lehner, K.R.; Thorndike, E.B.; Hoffman, A.F.; Holy, M.; Rothman, R.B.; Goldberg, S.R.; Lupica, C.R.; Sitte, H.H.; et al. Powerful Cocaine-like Actions of 3,4-Methylenedioxypyrovalerone (MDPV), a Principal Constituent of Psychoactive “Bath Salts” Products. Neuropsychopharmacology 2013, 38, 552–562. [Google Scholar] [CrossRef] [Green Version]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN Data Model for NMR Spectroscopy: Development of a Software Pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Rieping, W.; Habeck, M.; Bardiaux, B.; Bernard, A.; Malliavin, T.E.; Nilges, M. ARIA2: Automated NOE Assignment and Data Integration in NMR Structure Calculation. Bioinformatics 2007, 23, 381–382. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Bax, A. Protein Backbone and Sidechain Torsion Angles Predicted from NMR Chemical Shifts Using Artificial Neural Networks. J. Biomol. NMR 2013, 56, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, D.A.; Kaas, Q.; Rosengren, K.J. Prediction of Disulfide Dihedral Angles Using Chemical Shifts. Chem. Sci. 2018, 9, 6548–6556. [Google Scholar] [CrossRef] [Green Version]
- Nilges, M.; Bernard, A.; Bardiaux, B.; Malliavin, T.; Habeck, M.; Rieping, W. Accurate NMR Structures through Minimization of an Extended Hybrid Energy. Structure 2008, 16, 1305–1312. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for Checking the Quality of Protein Structures Solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 1, 12–21. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| χ-Conotoxin | Mature Sequence | IC50 NET | Cystine Connectivity | Ref. |
|---|---|---|---|---|
| PnID (Ribbon form) | STCCGYRMCVPC * | 10 μM | 1–4 & 2–3 | This work |
| MrIA | NGVCCGYKLCHOC * | 645 nM | 1–4 & 2–3 | [9,12,18] |
| MrIB | VGVCCGYKLCHOC * | 860 nM | 1–4 & 2–3 | [7] |
| CmrVIA | VCCGYKLCHOC * | N/A | 1–4 & 2–3 | [12] |
| PnID A | PnID B | |||
|---|---|---|---|---|
| Physical parameters | ||||
| Number of residues | 12 | 12 | ||
| Average molecular weight (unlabeled, Da) | 1317.6 | 1317.6 | ||
| Structural restraints | ||||
| NOE-derived distance restraints (ARIA cycle 8) | ||||
| Intraresidue (| I − j | = 0) | 90 | 72 | ||
| Sequential (| i − j | = 1) | 85 | 42 | ||
| Short (2 ≤ | i − j | ≤ 3) | 13 | 7 | ||
| Medium (4 ≤ | i − j | ≤ 5) | 4 | 1 | ||
| Long (| i − j | > 5) | 13 | 8 | ||
| Ambiguous | 29 | 15 | ||
| Total | 234 | 145 | ||
| Chemical shift-based dihedral constraints | ||||
| φ (from TALOS-N) | 6 | 5 | ||
| ψ (from TALOS-N) | 6 | 5 | ||
| Cystine χ1 and χ2 (from DISH) | 3 | 5 | ||
| Scalar coupling backbone torsion restraints (3JHNHA) | 9 | 0 | ||
| Hydrogen-bond restraints | 1 | 4 | ||
| Disulfide bond restraints | 2 | 2 | ||
| Statistics for accepted structures | ||||
| Accepted structures | 20 of 100 | 20 of 100 | ||
| Mean CNS energy terms | ||||
| E total (kcal mol−1) | −324 (±9) | −341(±12) | ||
| E van der Waals (kcal mol−1) | −33.3 (±1.2) | −32.3 (±1.8) | ||
| E distance restraints (kcal mol−1) | 70.03 (±0.06) | 44.013 (±0.002) | ||
| Restraint violations (average # per structure) | ||||
| NOE (>0.5 Å) | 0.4 (±0.6) | 2.2 (±1.2) | ||
| Dihedral (>5°) | 0 | 0 | ||
| 3JHNHA (>1 Hz) | 1.1 (±0.2) | N/A | ||
| RMS deviations from the ideal geometry used within CNS | ||||
| Bond lengths (Å) | 3.25 × 10−3 (±1.5 × 10−4) | 3.13 × 10−3 (±1.7 × 10−4) | ||
| Bond angles (°) | 0.41 (±0.02) | 0.36 (±0.02) | ||
| Improper angles (°) | 1.3 (±0.2) | 1.3 (±0.2) | ||
| Dihedral angles (°) | 39.8 (±0.3) | 41.0 (±0.7) | ||
| Ramachandran statistics (PROCHECK 3.5.4, [31]) | ||||
| Most favored (%) | 99.4 (±2.8) | 86.9 (±2.8) | ||
| Additionally allowed (%) | 0.6 (±2.8) | 10.6 (±4.6) | ||
| Generously allowed (%) | 0 | 2.5 (±5.1) | ||
| Disallowed (%) | 0 | 0 | ||
| MolProbity analyses (v3.19, [32]) | ||||
| Clashscore | 1.2 (±2.4) | 4.7 (±3.1) | ||
| Clashscore percentile (%) | 98.2 (±3.7) | 91.9 (±7.7) | ||
| Average atomic RMS deviations from average structure (±SD) | ||||
| N, Cα, C, and O atoms (all residues, Å) | 0.23 (±0.11) | 0.38 (±0.09) | ||
| All heavy atoms (all residues, Å) | 0.60 (±0.17) | 1.02 (±0.28) | ||
| N, Cα, C, and O atoms (for residues 2-11, Å) | 0.17 (±0.06) | 0.30 (±0.10) | ||
| All heavy atoms (for residues 2-11, Å) | 0.60 (±0.17) | 1.02 (±0.30) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espiritu, M.J.; Taylor, J.K.; Sugai, C.K.; Thapa, P.; Loening, N.M.; Gusman, E.; Baoanan, Z.G.; Baumann, M.H.; Bingham, J.-P. Characterization of the Native Disulfide Isomers of the Novel χ-Conotoxin PnID: Implications for Further Increasing Conotoxin Diversity. Mar. Drugs 2023, 21, 61. https://doi.org/10.3390/md21020061
Espiritu MJ, Taylor JK, Sugai CK, Thapa P, Loening NM, Gusman E, Baoanan ZG, Baumann MH, Bingham J-P. Characterization of the Native Disulfide Isomers of the Novel χ-Conotoxin PnID: Implications for Further Increasing Conotoxin Diversity. Marine Drugs. 2023; 21(2):61. https://doi.org/10.3390/md21020061
Chicago/Turabian StyleEspiritu, Michael J., Jonathan K. Taylor, Christopher K. Sugai, Parashar Thapa, Nikolaus M. Loening, Emma Gusman, Zenaida G. Baoanan, Michael H. Baumann, and Jon-Paul Bingham. 2023. "Characterization of the Native Disulfide Isomers of the Novel χ-Conotoxin PnID: Implications for Further Increasing Conotoxin Diversity" Marine Drugs 21, no. 2: 61. https://doi.org/10.3390/md21020061