
Chilensosides E, F, and G—New Tetrasulfated Triterpene Glycosides from the Sea Cucumber Paracaudina chilensis (Caudinidae, Molpadida): Structures, Activity, and Biogenesis
 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
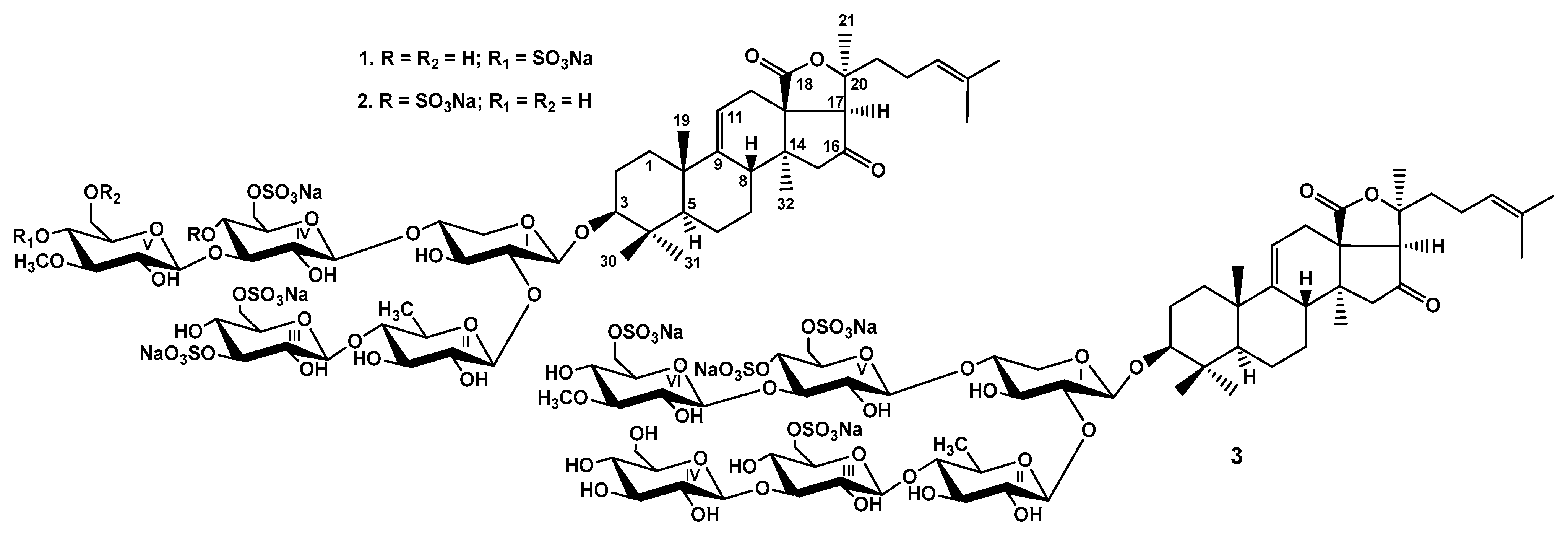
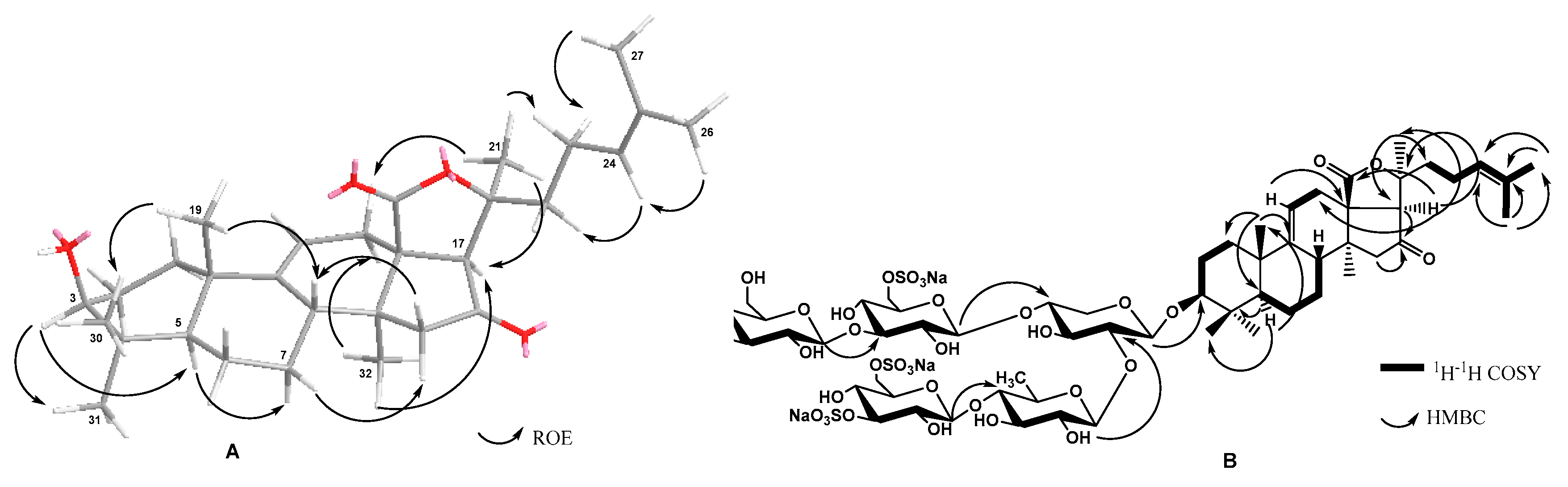
2.1. Structure Elucidation of the Glycosides
2.2. Bioactivity of the Glycosides
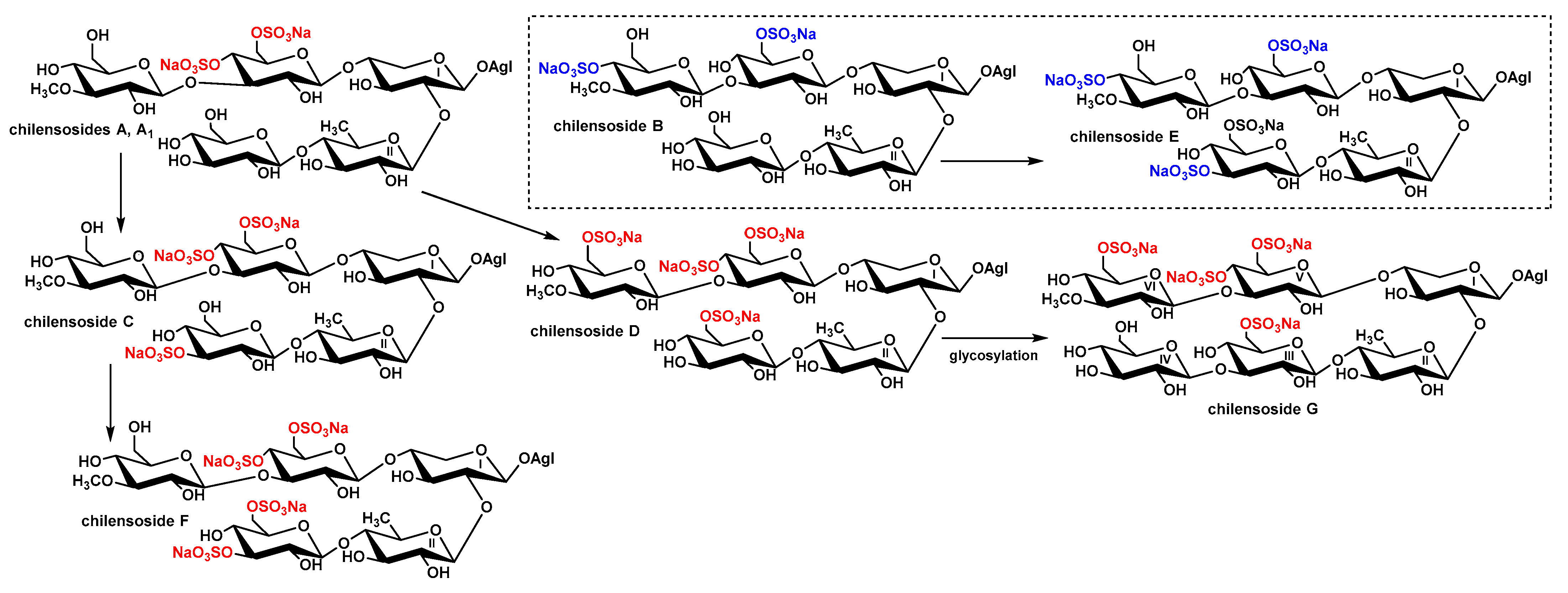
2.3. Biogenesis of Chilensosides A–G
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Animals and Cells
3.3. Extraction and Isolation
3.3.1. Chilensoside E (1)
3.3.2. Chilensoside F (2)
3.3.3. Chilensoside G (3)
3.4. Cytotoxic Activity (MTT Assay) (for SH-SY5Y, HeLa, and DLD-1 Cells)
3.5. Cytotoxic Activity (MTS Assay) (for HL-60 and THP-1 Cells)
3.6. Hemolytic Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; Stonik, V.A. Progress in the studies of triterpene glycosides from sea cucumbers (Holothuroidea, Echinodermata) between 2017 and 2021. Nat. Prod. Commun. 2021, 16, 10. [Google Scholar] [CrossRef]
- Mondol, M.A.M.; Shin, H.J.; Rahman, M.A. Sea cucumber glycosides: Chemical structures, producing species and important biological properties. Mar. Drugs 2017, 15, 317. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-I.; Bae, H.-R.; Kim, C.G.; Stonik, V.A.; Kwak, J.Y. Relationships between chemical structures and functions of triterpene glycosides isolated from sea cucumbers. Front. Chem. 2014, 2, 77. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.S. Biological activities of sulfated glycosides from Echinoderms. In Studies in Natural Product Chemistry (Bioactive Natural Products); Rahman, A.U., Ed.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 35, pp. 311–354. [Google Scholar]
- Khotimchenko, Y. Pharmacological potential of sea cucumbers. Int. J. Mol. Sci. 2020, 19, 1342. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-C.; Xue, C.-H.; Zhang, T.T.; Wang, Y.-M. Saponins from sea cucumber and their biological activities. Agric. Food Chem. 2018, 66, 7222–7237. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.R.; Freitas, A.C.; Duarte, A.C.; Rocha-Santos, T.A.P. Echinoderms: A review of bioactive compounds with potential health effects. In Studies in Natural Products Chemistry; Rahman, A.U., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 49, pp. 1–54. [Google Scholar]
- Chludil, H.D.; Murray, A.P.; Seldes, A.M.; Maier, M.S. Biologically active triterpene glycosides from sea cucumbers (Holothurioidea, Echinodermata). In Studies in Natural Products Chemistry; Rahman, A.U., Ed.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 28, pp. 587–616. [Google Scholar]
- Zelepuga, E.A.; Silchenko, A.S.; Avilov, S.A.; Kalinin, V.I. Structure-activity relationships of holothuroid’s triterpene glycosides and some in silico insights obtained by molecular dynamics study on the mechanisms of their membranolytic action. Mar. Drugs 2021, 19, 604. [Google Scholar] [CrossRef] [PubMed]
- Honey-Escandon, M.; Arreguin-Espinosa, R.; Solis-Martin, F.A.; Samyn, Y. Biological and taxonomic perspective of triterpenoid glycosides of sea cucumbers of the family Holothuriidae (Echinodermata, Holothuroidea). Comp. Biochem. Physiol. 2015, 180B, 16–39. [Google Scholar] [CrossRef] [PubMed]
- Omran, N.E.; Salem, ·H.K.; Eissa, S.H.; Kabbash, A.M.; Kandeil, ·M.A.; Salem, M.A. Chemotaxonomic study of the most abundant Egyptian sea-cucumbers using ultra-performance liquid chromatography (UPLC) coupled to high-resolution mass spectrometry (HRMS). Chemoecology 2020, 30, 35–48. [Google Scholar] [CrossRef]
- Kalinin, V.I.; Avilov, S.A.; Silchenko, A.S.; Stonik, V.A. Triterpene glycosides of sea cucumbers (Holothuroidea, Echinodermata) as taxonomic markers. Nat. Prod. Commun. 2015, 10, 21–26. [Google Scholar] [CrossRef]
- Smirnov, A.V. System of the class Holothuroidea. Paleontol. J. 2012, 46, 793–832. [Google Scholar] [CrossRef]
- Miller, A.K.; Kerr, A.M.; Paulay, G.; Reich, M.; Wilson, N.G.; Carvajal, J.I.; Rouse, G.W. Molecular phylogeny of extant Holothuroidea (Echinodermata). Mol. Phylogenet. Evol. 2017, 111, 110–131. [Google Scholar] [CrossRef] [PubMed]
- Silchenko, A.S.; Avilov, S.A.; Andrijaschenko, P.V.; Popov, R.S.; Chingizova, E.A.; Grebnev, B.B.; Rasin, A.B.; Kalinin, V.I. The isolation, structure elucidation and bioactivity study of chilensosides A, A1, B, C, and D, holostane triterpene di-, tri- and tetrasulfated pentaosides from the sea cucumber Paracaudina chilensis (Caudinidae, Molpadida). Molecules 2022, 27, 7655. [Google Scholar] [CrossRef] [PubMed]
- Silchenko, A.S.; Kalinovsky, A.I.; Avilov, S.A.; Kalinin, V.I.; Andrijaschenko, P.V.; Dmitrenok, P.S.; Popov, R.S.; Chingizova, E.A. Structures and bioactivities of psolusosides B1, B2, J, K, L, M, N, O, P, and Q from the sea cucumber Psolus fabricii. The first finding of tetrasulfated marine low molecular weight metabolites. Mar. Drugs 2019, 17, 631. [Google Scholar] [CrossRef] [PubMed]
- Silchenko, A.S.; Avilov, S.A.; Andrijaschenko, P.V.; Popov, R.S.; Chingizova, E.A.; Dmitrenok, P.S.; Kalinovsky, A.I.; Rasin, A.B.; Kalinin, V.I. Structures and biologic activity of chitonoidosides I, J, K, K1 and L, triterpene di-, tri- and tetrasulfated hexaosides from the sea cucumber Psolus chitonoides. Mar. Drugs 2022, 20, 369. [Google Scholar] [CrossRef] [PubMed]
- Silchenko, A.S.; Avilov, S.A.; Kalinin, V.I. Separation procedures for complicated mixtures of sea cucumber triterpene glycosides with isolation of individual glycosides, their comparison with HPLC/MS metabolomic approach, and biosynthetic interpretation of the obtained structural data. In Studies in Natural Product Chemistry; Rahman, A.U., Ed.; Elsevier: Amsterdam, The Netherlands, 2022; Volume 72, pp. 103–146. [Google Scholar] [CrossRef]
- Aminin, D.L.; Menchinskaya, E.S.; Pislyagin, E.A.; Silchenko, A.S.; Avilov, S.A.; Kalinin, V.I. Sea cucumber triterpene glycosides as anticancer agents. In Studies in Natural Product Chemistry; Rahman, A.U., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 49, pp. 55–105. [Google Scholar]
- Kalinin, V.I.; Aminin, D.L.; Avilov, S.A.; Silchenko, A.S.; Stonik, V.A. Triterpene glycosides from sea cucucmbers (Holothurioidae, Echinodermata), biological activities and functions. In Studies in Natural Product Chemistry (Bioactive Natural Products); Rahman, A.U., Ed.; Elsevier: The Netherlands, 2008; Volume 35, pp. 135–196. [Google Scholar]
- Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; Stonik, V.A. Non-holostane aglycones of sea cucumber triterpene glycosides. Structure, biosynthesis, evolution. Steroids 2019, 147, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, S.; Arakawa, O.; Terada, M.; Nishio, S.; Takatani, T.; Mahmud, Y.; Noguchi, T. Ostreopsis sp., a possible origin of palytoxin (PTX) in parrotfish Scarus ovifrons. Toxicon 2003, 42, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Malagoli, D.A. Full-Length Protocol to Test Hemolytic Activity of Palytoxin on Human Erythrocytes; Technical Report; Department of Animal Biology, University of Modena and Reggio Emilia: Modena, Italy, 2007; pp. 92–94. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Position | δC Mult. a | δH Mult. (J in Hz) b | HMBC | ROESY |
|---|---|---|---|---|
| 1 | 36.0 CH2 | 1.73 m | H-11 | |
| 1.30 m | H-3 | |||
| 2 | 26.8 CH2 | 2.06 m | ||
| 1.84 m | H-19, H-30 | |||
| 3 | 88.4 CH | 3.11 dd (4.7; 11.8) | H-1, H-5, H-31, H1-Xyl1 | |
| 4 | 39.5 C | |||
| 5 | 52.7 CH | 0.79 brd (11.8) | C: 4, 19, 30 | H-1, H-3, H-7 |
| 6 | 20.9 CH2 | 1.59 m | ||
| 7 | 28.3 CH2 | 1.60 m | H-15 | |
| 1.12 m | ||||
| 8 | 38.6 CH | 3.14 m | H-6 | |
| 9 | 151.1 C | |||
| 10 | 39.5 C | |||
| 11 | 111.3 CH | 5.27 brs | C: 10, 13 | H-1 |
| 12 | 31.9 CH2 | 2.64 brd (16.5) | C: 11, 18 | H-17 |
| 2.48 dd (5.9; 16.5) | C: 11, 14 | |||
| 13 | 56.0 C | |||
| 14 | 42.0 C | |||
| 15 | 51.8 CH2 | 2.42 d (16.0) | C: 13, 16, 17, 32 | |
| 2.11 d (16.0) | C: 14, 16, 32 | H-8 | ||
| 16 | 214.6 C | |||
| 17 | 61.8 CH | 2.89 s | C: 12, 13, 16, 18, 20, 21 | H-12, H-23, H-32 |
| 18 | 176.9 C | |||
| 19 | 21.9 CH3 | 1.27 s | C: 1, 5, 9, 10 | H-1, H-2, H-8, H-30 |
| 20 | 83.1 C | |||
| 21 | 26.6 CH3 | 1.48 s | C: 17, 20, 22 | H-12, H-17, H-23 |
| 22 | 38.6 CH2 | 1.80 m | ||
| 1.59 m | ||||
| 23 | 23.0 CH2 | 2.27 m | ||
| 2.08 m | ||||
| 24 | 124.1 CH | 5.03 m | H-22 | |
| 25 | 132.1 C | |||
| 26 | 25.5 CH3 | 1.56 s | C: 24, 25, 27 | H-24 |
| 27 | 17.4 CH3 | 1.54 s | C: 24, 25, 26 | H-23 |
| 30 | 16.4 CH3 | 0.89 s | C: 3, 4, 5, 31 | H-2, H-6, H-19, H-31 |
| 31 | 27.8 CH3 | 1.10 s | C: 3, 4, 5, 30 | H-3, H-5, H-6, H-30 |
| 32 | 20.5 CH3 | 0.89 s | C: 8, 13, 14, 15 | H-7, H-12, H-15, H-17 |
| Atom | δC Mult. a,b,c | δH Mult. (J in Hz) d | HMBC | ROESY |
|---|---|---|---|---|
| Xyl1 (1→C-3) | ||||
| 1 | 104.6 CH | 4.61 d (7.4) | C: 3 | H-3; H-5 Xyl1 |
| 2 | 83.4 CH | 3.70 m | C: 3 Xyl1 | H-1 Qui2 |
| 3 | 75.0 CH | 3.93 m | ||
| 4 | 80.8 CH | 3.94 m | C: 3 Xyl1; 1 Glc4 | H-1 Glc4; H-2 Xyl1 |
| 5 | 63.4 CH2 | 4.33 dd (6.0; 12.3) | ||
| 3.58 m | H-1 Xyl1 | |||
| Qui2 (1→2Xyl1) | ||||
| 1 | 104.6 CH | 4.74 d (8.3) | C: 2 Xyl1 | H-2 Xyl1; H-3, 5 Qui2 |
| 2 | 75.5 CH | 3.92 t (9.4) | C: 3 Qui2 | H-4 Qui2 |
| 3 | 74.4 CH | 4.06 t (9.4) | H-1, 5 Qui2 | |
| 4 | 86.0 CH | 3.30 t (9.4) | C: 1 Glc3 | H-1 Glc3 |
| 5 | 71.7 CH | 3.60 dd (6.0; 9.4) | H-1, 3 Qui2 | |
| 6 | 17.4 CH3 | 1.54 d (6.0) | C: 4, 5 Qui2 | H-4 Qui2 |
| Glc3 (1→4Qui2) | ||||
| 1 | 104.1 CH | 4.67 d (7.9) | C: 4 Qui2 | H-4 Qui2; H-3, 5 Glc3 |
| 2 | 72.8 CH | 3.81 t (7.9) | C: 1, 3 Glc3 | |
| 3 | 83.8 CH | 4.98 t (7.9) | H-1, 5 Glc3 | |
| 4 | 69.5 CH | 3.80 t (7.9) | C: 3, 5 Glc3 | H-6 Glc3 |
| 5 | 74.2 CH | 4.07 t (7.9) | ||
| 6 | 67.4 CH2 | 4.91 brd (9.8) | ||
| 4.52 dd (7.3; 11.0) | ||||
| Glc4 (1→4Xyl1) | ||||
| 1 | 103.4 CH | 4.85 d (7.9) | C: 4 Xyl1 | H-4 Xyl1; H-3, 5 Glc4 |
| 2 | 74.0 CH | 3.83 t (7.9) | ||
| 3 | 85.8 CH | 4.17 t (9.2) | C: 1 MeGlc5 | H-1 MeGlc5; H-1 Glc4 |
| 4 | 69.5 CH | 3.68 t (9.2) | C: 5 Glc4 | |
| 5 | 74.4 CH | 4.16 m | ||
| 6 | 67.7 CH2 | 4.95 brd (10.5) | ||
| 4.44 brdd (7.2; 11.2) | ||||
| MeGlc5 (1→3Glc4) | ||||
| 1 | 104.3 CH | 5.16 d (8.3) | C: 3 Glc4 | H-3 Glc4; H-5 MeGlc5 |
| 2 | 74.0 CH | 3.87 t (8.3) | C: 1 MeGlc5 | H-4 MeGlc5 |
| 3 | 85.3 CH | 3.71 t (8.3) | C: 4 MeGlc5; OMe | H-1 Me Glc5 |
| 4 | 76.1 CH | 4.88 t (8.8) | C: 5 MeGlc5 | |
| 5 | 76.4 CH | 3.86 m | H-1 MeGlc5 | |
| 6 | 61.7 CH2 | 4.49 d (11.6) | ||
| 4.33 m | ||||
| OMe | 60.8 CH3 | 3.93 s | C: 3 MeGlc5 | H-3 MeGlc5 |
| Atom | δC Mult. a,b,c | δH Mult. (J in Hz) d | HMBC | ROESY |
|---|---|---|---|---|
| Xyl1 (1→C-3) | ||||
| 1 | 104.6 CH | 4.62 d (7.7) | C: 3 | H-3; H-3, 5 Xyl1 |
| 2 | 82.9 CH | 3.71 t (7.7) | C: 1, 3 Xyl1 | H-1 Qui2 |
| 3 | 75.1 CH | 3.93 t (7.7) | C: 2, 4 Qui2 | H-1 Xyl1 |
| 4 | 81.2 CH | 3.92 m | C: 1 Glc4 | H-1 Glc4 |
| 5 | 63.4 CH2 | 4.28 dd (5.1; 11.5) | C: 3 Xyl1 | |
| 3.57 m | H-1, 3 Xyl1 | |||
| Qui2 (1→2Xyl1) | ||||
| 1 | 104.7 CH | 4.75 d (7.3) | C: 2 Xyl1 | H-2 Xyl1; H-3, 5 Qui2 |
| 2 | 75.3 CH | 3.94 t (9.3) | H-4 Qui2 | |
| 3 | 74.4 CH | 4.06 t (9.3) | C: 2, 4 Qui2 | H-1, 5 Qui2 |
| 4 | 85.9 CH | 3.31 t (9.3) | C: 1 Glc3; 5 Qui2 | H-1 Glc3; H-2 Qui2 |
| 5 | 71.7 CH | 3.60 m | H-1, 3 Qui2 | |
| 6 | 17.4 CH3 | 1.53 d (6.4) | C: 4, 5 Qui2 | H-4 Qui2 |
| Glc3 (1→4Qui2) | ||||
| 1 | 104.1 CH | 4.68 d (7.5) | C: 4 Qui2 | H-4 Qui2; H-3, 5 Glc3 |
| 2 | 72.8 CH | 3.80 t (9.0) | C: 1, 3 Glc3 | |
| 3 | 83.9 CH | 4.98 t (9.0) | C: 2, 4 Glc3 | H-1, 5 Glc3 |
| 4 | 69.5 CH | 3.82 t (9.0) | C: 3, 5, 6 Glc3 | |
| 5 | 74.3 CH | 4.08 t (9.0) | H-1, 3 Glc3 | |
| 6 | 67.4 CH2 | 4.92 brd (11.7) | ||
| 4.53 dd (7.6; 11.7) | C: 5 Glc3 | H-4 Glc3 | ||
| Glc4 (1→4Xyl1) | ||||
| 1 | 103.5 CH | 4.81 d (7.8) | C: 4 Xyl1 | H-4 Xyl1; H-3, 5 Glc4 |
| 2 | 74.0 CH | 3.92 t (8.4) | C: 1, 3 Glc4 | |
| 3 | 82.8 CH | 4.37 t (8.4) | C: 1 MeGlc5; 2, 4 Glc4 | H-1 MeGlc5 |
| 4 | 75.5 CH | 4.74 t (8.4) | C: 5, 6 Glc4 | H-2 Glc4 |
| 5 | 73.8 CH | 4.33 m | H-1 Glc4 | |
| 6 | 68.5 CH2 | 5.50 m | ||
| 4.62 t (10.9) | H-4 Glc4 | |||
| MeGlc5 (1→3Glc4) | ||||
| 1 | 104.5 CH | 5.20 d (8.2) | C: 3 Glc4 | H-3 Glc4; H-3, 5 MeGlc5 |
| 2 | 74.5 CH | 4.00 t (9.4) | C: 1, 3 MeGlc5 | |
| 3 | 86.9 CH | 3.65 t (9.4) | C: 2, 4 MeGlc5; OMe | H-1, 5 Me Glc5; OMe |
| 4 | 70.0 CH | 3.91 t (9.4) | C: 3, 5, 6 MeGlc5 | H-6 MeGlc5 |
| 5 | 77.5 CH | 3.87 m | H-1 MeGlc5 | |
| 6 | 62.0 CH2 | 4.34 brd (11.7) | ||
| 4.09 dd (7.0; 11.7) | C: 5 MeGlc5 | |||
| OMe | 60.3 CH3 | 3.76 s | C: 3 MeGlc5 |
| Atom | δC Mult. a,b,c | δH Mult. (J in Hz) d | HMBC | ROESY |
|---|---|---|---|---|
| Xyl1 (1→C-3) | ||||
| 1 | 104.7 CH | 4.65 d (6.8) | C: 3 | H-3; H-3, 5 Xyl1 |
| 2 | 82.2 CH | 3.85 t (8.7) | C: 1 Xyl1 | H-1 Qui2 |
| 3 | 75.0 CH | 4.10 t (8.7) | C: 2, 4 Qui2 | H-5 Xyl1 |
| 4 | 79.5 CH | 4.05 m | H-1 Glc5 | |
| 5 | 63.4 CH2 | 4.34 dd (4.9; 11.7) | C: 3 Xyl1 | |
| 3.63 brdd (8.7; 11.7) | H-1 Xyl1 | |||
| Qui2 (1→2Xyl1) | ||||
| 1 | 104.5 CH | 4.89 d (8.0) | C: 2 Xyl1 | H-2 Xyl1; H-3, 5 Qui2 |
| 2 | 75.4 CH | 3.83 t (8.9) | C: 1, 3 Qui2 | H-4 Qui2 |
| 3 | 74.7 CH | 3.92 t (8.3) | C: 2 Qui2 | H-1 Qui2 |
| 4 | 86.9 CH | 3.34 t (8.3) | C: 1 Glc3; 3, 5 Qui2 | H-1 Glc3; H-2 Qui2 |
| 5 | 71.4 CH | 3.61 dd (6.2; 8.3) | H-1, 3 Qui2 | |
| 6 | 17.6 CH3 | 1.57 d (6.5) | C: 4, 5 Qui2 | H-4 Qui2 |
| Glc3 (1→4Qui2) | ||||
| 1 | 104.2 CH | 4.71 d (7.8) | C: 4 Qui2 | H-4 Qui2; H-3, 5 Glc3 |
| 2 | 73.4 CH | 3.83 t (8.6) | C: 1, 3 Glc3 | |
| 3 | 86.1 CH | 4.16 t (8.6) | C: 1 Glc4; 2, 4 Glc3 | H-1 Glc4; H-1 Glc3 |
| 4 | 69.3 CH | 3.75 t (8.6) | C: 3, 5, 6 Glc3 | H-6 Glc3 |
| 5 | 74.7 CH | 4.08 t (8.6) | H-1 Glc3 | |
| 6 | 67.4 CH2 | 4.95 brd (10.2) | C: 4 Glc3 | |
| 4.54 dd (7.1; 11.0) | H-4 Glc3 | |||
| Glc4 (1→3Glc3) | ||||
| 1 | 104.6 CH | 5.19 d (7.8) | C: 3 Glc3 | H-3 Glc3; H-3, 5 Glc4 |
| 2 | 74.9 CH | 3.91 t (8.6) | C: 3 Glc4 | |
| 3 | 77.2 CH | 4.09 t (8.6) | C: 2, 4 Glc4 | H-1 Glc4 |
| 4 | 71.0 CH | 3.88 t (8.6) | ||
| 5 | 77.7 CH | 3.90 t (8.6) | H-1 Glc4 | |
| 6 | 61.9 CH2 | 4.35 d (11.8) | ||
| 4.05 dd (5.5; 11.8) | C: 5 Glc4 | |||
| Glc5 (1→4Xyl1) | ||||
| 1 | 102.7 CH | 4.81 d (8.7) | C: 4 Xyl1 | H-4 Xyl1; H-3, 5 Glc5 |
| 2 | 73.5 CH | 3.96 t (8.7) | C: 1, 3 Glc5 | |
| 3 | 80.1 CH | 4.52 t (8.7) | C: 1 MeGlc6; 2, 4 Glc5 | H-1 MeGlc6; H-1, 5 Glc5 |
| 4 | 75.3 CH | 4.76 t (8.7) | C: 3, 5, 6 Glc5 | H-6 Glc5 |
| 5 | 73.8 CH | 4.25 t (8.7) | C: 4, 6 Glc5 | H-1, 3 Glc5 |
| 6 | 68.2 CH2 | 5.39 d (10.9) | ||
| 4.66 dd (7.6; 10.9) | C: 5 MeGlc5 | |||
| MeGlc6 (1→3Glc5) | ||||
| 1 | 102.3 CH | 5.38 d (7.8) | C: 3 Glc5 | H-3 Glc5; H-3, 5 MeGlc6 |
| 2 | 74.0 CH | 3.94 t (7.8) | C: 1, 3 MeGlc6 | |
| 3 | 86.3 CH | 3.61 t (8.9) | OMe; C: 2, 4 MeGlc6 | OMe; H-1, 5 MeGlc6 |
| 4 | 69.4 CH | 4.04 t (8.9) | C: 3, 5, 6 MeGlc6 | |
| 5 | 75.6 CH | 3.97 m | H-1, 3 MeGlc6 | |
| 6 | 66.8 CH2 | 4.94 d (10.4) | C: 4 MeGlc6 | |
| 4.79 dd (5.2; 11.9) | ||||
| OMe | 60.3 CH3 | 3.73 s | C: 3 MeGlc6 |
| Glycosides | ED50, µM, Erythrocytes | Cytotoxicity, IC50 µM | ||||
|---|---|---|---|---|---|---|
| SH-SY5Y | HeLa | DLD-1 | HL-60 | THP-1 | ||
| Chilensoside E (1) | 29.89 ± 2.67 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 |
| Chilensoside F (2) | 34.31 ± 0.63 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 |
| Chilensoside G (3) | 40.74 ± 1.55 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 |
| Chitonoidoside L | 1.12 ± 0.10 | 8.06 ± 0.98 | 14.36 ± 1.12 | 9.61 ± 1.24 | 8.22 ± 0.65 | 8.32 ± 0.81 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silchenko, A.S.; Avilov, S.A.; Popov, R.S.; Dmitrenok, P.S.; Chingizova, E.A.; Grebnev, B.B.; Rasin, A.B.; Kalinin, V.I. Chilensosides E, F, and G—New Tetrasulfated Triterpene Glycosides from the Sea Cucumber Paracaudina chilensis (Caudinidae, Molpadida): Structures, Activity, and Biogenesis. Mar. Drugs 2023, 21, 114. https://doi.org/10.3390/md21020114
Silchenko AS, Avilov SA, Popov RS, Dmitrenok PS, Chingizova EA, Grebnev BB, Rasin AB, Kalinin VI. Chilensosides E, F, and G—New Tetrasulfated Triterpene Glycosides from the Sea Cucumber Paracaudina chilensis (Caudinidae, Molpadida): Structures, Activity, and Biogenesis. Marine Drugs. 2023; 21(2):114. https://doi.org/10.3390/md21020114
Chicago/Turabian StyleSilchenko, Alexandra S., Sergey A. Avilov, Roman S. Popov, Pavel S. Dmitrenok, Ekaterina A. Chingizova, Boris B. Grebnev, Anton B. Rasin, and Vladimir I. Kalinin. 2023. "Chilensosides E, F, and G—New Tetrasulfated Triterpene Glycosides from the Sea Cucumber Paracaudina chilensis (Caudinidae, Molpadida): Structures, Activity, and Biogenesis" Marine Drugs 21, no. 2: 114. https://doi.org/10.3390/md21020114