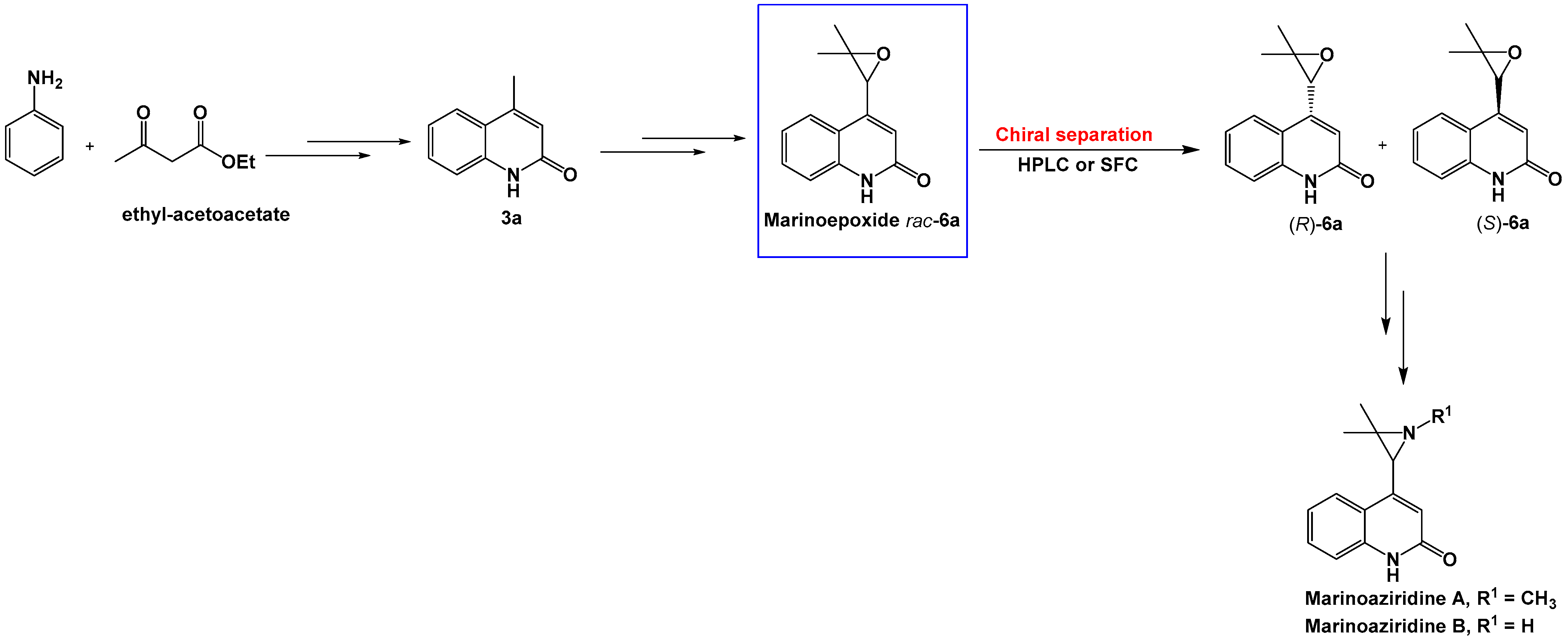
In this study, chromatographic conditions for the separation of marinoepoxides enantiomers
rac-
6a–
c and
rac-8a–
c on immobilized chiral stationary phases were investigated using high-performance liquid chromatography (HPLC) and supercritical fluid chromatography (SFC). Three chiral columns, CHIRAL ART Cellulose-SC, CHIRAL ART Cellulose-SB and CHIRAL ART Amylose-SA with immobilized chiral selector were examined (
Table 2). In our preliminary studies, the CHIRAL ART Cellulose-SB column did not prove to be a good choice in the separation of marinoepoxides enantiomers, so the optimization of chromatographic conditions was performed on the remaining two immobilized stationary phases. The developed enantioselective analytical methods will be used in control of enantiomeric purity during stereoselective synthesis of target molecules, and also on a preparative scale chiral chromatography. Chiral recognition using polysaccharide-based chiral stationary phases is based on a complex process involving chiral and achiral interactions between the analyte, the chiral selector and the mobile phase. Enantiorecognition of trisubstituted
rac-
6a–
c and disubstituted marinoepoxides
rac-
8a–
c is based on attractive interactions such as π-π interactions, dipole-dipole interactions and hydrogen bonds, and repulsive interactions that are usually steric. Polar and π-π interactions between phenyl groups of the chiral stationary phase and the analyte play an important role in the chiral recognition mechanism [
24,
25,
26] Based on numerous studies, it is difficult to predict which chiral selector of a particular stationary phase will separate the enantiomers of a compound, and which mobile phase will be the most favorable. The development of a good enantioselective chromatographic method requires finding the best combination of process parameters, described by chiral stationary phase selection, mobile phase composition and concentration, column temperature and back pressure (mainly for SFC) [
27].
All investigated marinoepoxides
rac-
6a–
c, and
rac-
8a–
c have different substitutions (steric effects) on the nitrogen atom of the internal amide bond of quinoline-2(1
H)-one moiety, and therefore possess different spatial orientation, due to which they possess different affinities towards the chiral stationary phase selector. In the case of disubstituted marinoepoxides
rac-
8a–
c, it was important to determine whether the bulkiness of the phenyl ring on the C3 carbon atom of the epoxy ring (inability to adhere to the chiral cavity) or π-π interactions with the stationary phase selector is decisive for chiral recognition. Marinoepoxides
rac-
6a–
c contain two methyl groups on the C3 atom instead of the phenyl ring, and the effect of less voluminous groups on the chiral recognition mechanism was investigated. The enantioseparation tests were firstly performed using HPLC with
n-hexane/ethanol (80/20,
v/
v) as a standard, widely used as a solvent system to compare with green alternatives. Further enantioseparation tests were performed using green solvent systems as pure dimethyl carbonate and dimethyl carbonate/alcohol (
v/
v) mixtures. Particular attention was paid to the use of SFC with CO
2/alcohol (
v/
v) as nonpolar mobile phase. In the
n-hexane binary system, only ethanol was used as the alcohol modifier. Using propan-2-ol, a long retention time of enantiomers (t
R > 110 min) was observed, while methanol was not used in this study due to poor miscibility with
n-hexane. The effect of the addition of an alcohol modifier (methanol or ethanol) on the enantioselectivity and retention times was investigated also in the dimethyl carbonate binary system as well as in the SFC system. It has been hypothesized that increasing the polarity of the mobile phase leads to less pronounced hydrogen bonds between the chiral stationary phase and the analyte. Moreover, alcohol molecules combine with the chiral stationary phase and cause swelling of the column, which directly affects the size and shape of the chiral cavity. If chiral cavities are opened, the inclusion interactions of the analyte are reduced and the retention time of the analyte is reduced [
24,
28,
29,
30]. Pirkle and Welch studied the effect of an alcohol modifier on enantioselectivity and found that the effect of a modifier depends on the structure of the analyte [
31,
32,
33]. Based on VCD measurements Grinberg et al. observed conformational changes of the chiral stationary phase in the presence of polar solvents. Conformations changed drastically depending on the concentration and effect of the shape and size of chiral cavities, i.e., the mechanism of chiral recognition [
34].
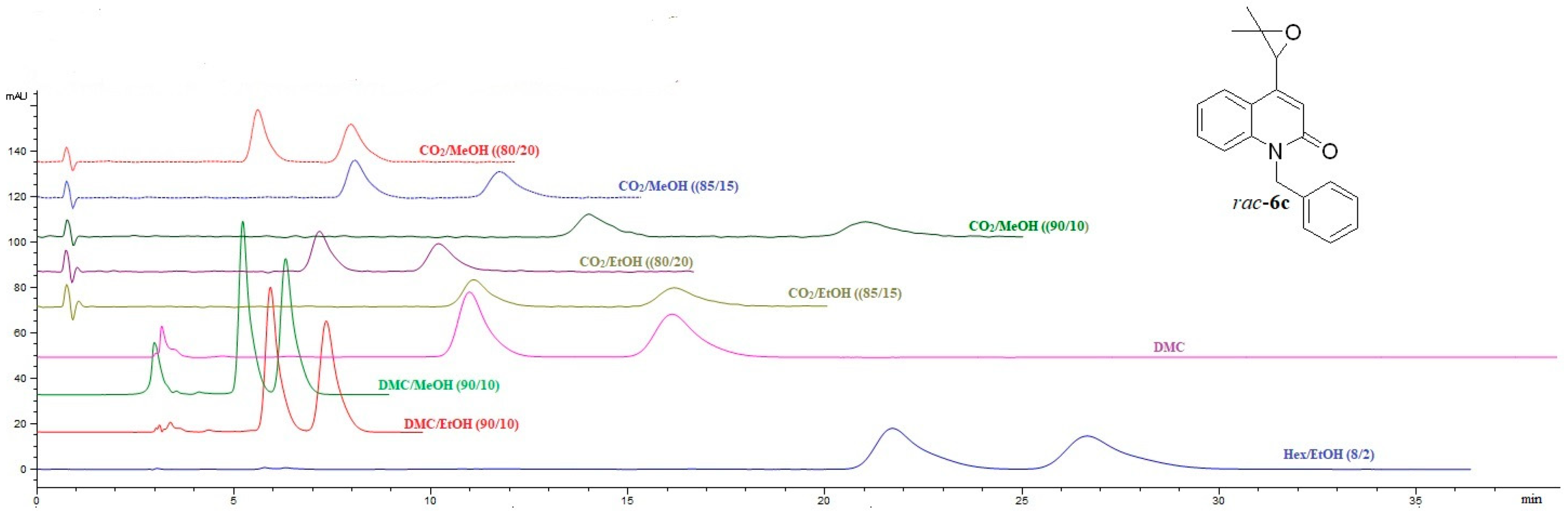
2.2.1. HPLC and SFC Enantioselective Chromatography of (±)-Trisubstituted Marinoepoxides rac-6a–c
The results of HPLC enantioselective analysis of marinoepoxides
rac-
6a–
c are shown in
Table S1 (Supplementary materials) (
Figure 3 and
Figure 4). It was observed that enantioseparation of marinoepoxides
rac-
6a,
b shows a higher degree of enantioselectivity on the CHIRAL ART Amylose-SA column, while compound
rac-
6c achieves better enantioselectivity on the CHIRAL ART Cellulose-SC column when using hexane/EtOH (80/20,
v/
v) as a mobile phase. By replacing hexane/EtOH (80/20,
v/
v) mobile phase with dimethyl carbonate, all enantiomers of marinoepoxides
rac-
6a–
c showed enantioseparation on the CHIRAL ART Cellulose-SC column in comparison to CHIRAL ART Amylose-SA column.
From the obtained results, excellent baseline enantioseparation of all marinoepoxides
rac-
6a–
c on the selected CHIRAL ART Cellulose-SC column in the HPLC mode was observed. Comparing the separation factors for enantiomers of all three compounds
rac-
6a–
c with
n-hexane/ethanol (80/20,
v/
v), it can be observed that the degree of enantioselectivity is almost independent of the substituent (-H, -CH
3, -Bn) on the nitrogen atom of the internal amide bond of quinoline-2(1
H)-one moiety. This finding indicates that the mechanism of chiral recognition is not dominated by steric interactions. The chiral stationary phase selector in CHIRAL ART Cellulose-SC contains two electron-withdrawing groups that reduce the electron density of the benzene system and increase the hydrogen acidity of the phenylcarbamate group [
35]. Given these facts, it can be assumed that chiral recognition of marinoepoxide enantiomers
rac-
6a–
c with selector
tris-(3,5-dichlorophenylcarbamoyl)cellulose is based on hydrogen bonds and dipole-dipole interactions. By comparing the retention factors, it can be observed that the enantiomers of compound
rac-
6b show the longest retention time on the tested cellulose column. The longer retention of the enantiomers of compound
rac-
6b on the column relative to the remaining two compounds
rac-
6a and
rac-
6c may be due to strong dipole-dipole interactions or hydrogen bonds that can further stabilize the diastereomeric complex [
36]. The electron-donating methyl group on the nitrogen atom in the analyte structure makes the C=O group a better acceptor of the hydrogen bond that can be achieved with the hydrogen atom in the phenylcarbamate group of the chiral selector. Longer retention times for enantiomers of
rac-
6b (
Rs = 2.67) led to higher values of the resolution factor compared to the enantiomers of compounds
rac-
6a (
Rs = 2.12) and
rac-
6c (
Rs = 2.13).
The use of dimethyl carbonate as a mobile phase reduces the retention time of the enantiomers and increases the resolution factor for all marinoepoxides rac-6a–c in the sequence: k1 (20.34; rac-6a) > k1 (19.15; rac-6b) > k1 (10.96; rac-6c), or Rs (4.70; rac-6b) > Rs (3.31; rac-6a) > Rs (3.19; rac-6c). Shorter retention of enantiomers and higher values of Rs resolution factors for compounds rac-6a–c may be the result of dipole-dipole interactions of the solvent and stationary phase, with these interactions contributing to more significant enantiorecognition of chiral phase and analytes. This result confirms the thesis that the contribution of π-π interactions in the process of chiral discrimination is very small. The addition of methanol or ethanol with a volume fraction of 10% to the dimethyl carbonate mobile phase increases the polarity of the mobile phase and reduces the retention factor of the analyte on the stationary phase in the range kH > kCH3 > kBn. This indicates a strong competition of alcohols and analytes for interaction sites on the chiral stationary phase. At a volume fraction of 10% methanol or ethanol, α values and resolution factor decrease.
If we compare the enantioselectivity for enantiomers of compounds
rac-
6a–
c on cellulose and amylose columns using SFC chromatography, then the enantiomers of compound
rac-
6a showed better enantioselectivity on the CHIRAL ART Amylose-SA column. The different chiral recognition mechanism is possibly due to the formation of different shapes and sizes of chiral cavities that build carbamate groups on adjacent glucose units of amylose and cellulose depending on the mobile phase [
37].
The application of supercritical fluid chromatography in the enantioseparation of marinoepoxides rac-6a–c on a selected cellulose column showed excellent chiral recognition with a stationary phase selector, regardless of the type and volume fraction of alcohol modifier in the CO2 binary system. All enantiomers of compounds rac-6a–c were separated to the baseline, regardless of the composition of the mobile phase. By comparing the retention factors of the starting enantiomers rac-6a–c on the cellulose column, it was observed that the enantiomers remain longer with ethanol as an alcohol modifier in the series kH > kCH3 > kBn. The separation factors of the enantiomers of compounds rac-6a–c are almost constant by changing the type and volume fraction of the alcohol modifier. Compound rac-6a (α = 1.62) showed the best enantioselectivity with CO2/EtOH (80/20, v/v) mobile phase, while compounds rac-6b (α = 1.58) and rac-6c (α = 1.51) showed the best enantioselectivity using CO2/MeOH (80/20, v/v) as mobile phase. The values of the separation factors differ slightly by changing the alcohol modifier. By comparing the separation factors in the HPLC and SFC mode, it was observed that compounds rac-6b and rac-6c show better enantioselectivity with dimethyl carbonate compared to the enantioselectivity obtained using CO2/alcohol (v/v) as binary mobile phase. Compound rac-6a showed better enantioselectivity using supercritical fluid chromatography, regardless of the composition and volume of the alcohol in the CO2 mobile phase. By comparing the resolution factors of the enantiomers of compounds rac-6a–c, excellent resolution (Rs > 2.06) was observed in the entire test range from 20% to 5% by volume of alcohol in the CO2 binary system. The presented results indicate that the reduction of the alcohol content by volume in the CO2 binary system leads to longer retention of the enantiomer on the column and better resolution, i.e., better enantiorecognition of the analyte with the stationary phase selector.
The main advantage of developed SFC enantioselective methods is particularly visible during HPLC and SFC enantioseparation of the key intermediate molecule and its derivatives, racemic marinoepoxides rac-6a–c, on CHIRAL ART Amylose-SA and CHIRAL ART Cellulose-SC columns. In all cases, SFC chiral chromatography proved to be the method of choice in terms of lower retention times and higher separation factors. This is especially true when using CHIRAL ART Amylose-SA for enantioseparation of rac-6b. In this case the only green chromatography method is achieved using SFC, while using HPLC with green solvent (DMC) revealed only partial enantioseparation. For comparison, the HPLC method using n-hexane/EtOH mixture revealed longer retention times and lower enantioselectivity for rac-6b.
Figure 4 shows the enantioseparation chromatograms of marinoepoxides
rac-
6a–
c separated on a CHIRAL ART Cellulose-SC column, with different mobile phase compositions, both in HPLC and SFC mode.
The amylose-based chiral stationary phase CHIRAL ART Amylose-SA contains two electron-donating methyl groups on the phenyl ring that increase the electron density of the benzene ring and the C=O group and reduce the hydrogen acidity of the phenylcarbamate group of the chiral selector. Given the described electronic properties of the amylose-based chiral stationary phase, enantiorecognition of trisubstituted marinoepoxides rac-6a–c is based on the formation of π-π interactions and dipole-dipole interactions, and repulsive interactions that are mostly steric. In the case of compound rac-6a, both the hydrogen bond via the C=O group of the chiral selector and the hydrogen atom of the internal amide bond of marinoepoxide structure can be achieved.
Using SFC chromatography in enantioseparation of marinoepoxides rac-6a–c, revealed an excellent baseline separation of rac-6a. The enantiomers of compound rac-6a are retained shorter with better resolution in the SFC mode compared to the HPLC mode. Moreover, the enantiomers of compound rac-6b showed excellent baseline separation using alcohol modifier ranging from 3% to 10% (methanol or ethanol, v/v). By comparing the retention factors of enantiomers rac-6a–c on the amylose-based column, it can be observed that the enantiomers retain longer with ethanol as modifier in the sequence kBn > kH < kCH3. The separation factors for enantiomers of rac-6a–c are almost constant by changing the type and volume fraction of the alcohol modifier. The highest values of the separation factors for compounds rac-6a–c were achieved for compound rac-6a (α = 2.12) with CO2/EtOH (80/20, v/v), compound rac-6b (α = 1.55) with CO2 / MeOH (97/3, v/v) and compound rac-6c (α = 1.25) with CO2/EtOH (95/5, v/v). The highest values of the resolution factor Rs in the SFC mode were achieved for compound rac-6a (Rs = 4.83) with CO2/EtOH (95/5, v/v), compound rac-6b (Rs = 3.01) with CO2/MeOH (97/3, v/v) and compound rac-6c (Rs = 2.04) with CO2/EtOH (95/5, v/v). The presented results indicate that the reduction of the alcohol content by volume in the CO2 binary system leads to longer retention of the enantiomers on the column and better resolution, i.e., better enantiorecognition of the analyte with the chiral stationary phase selector.
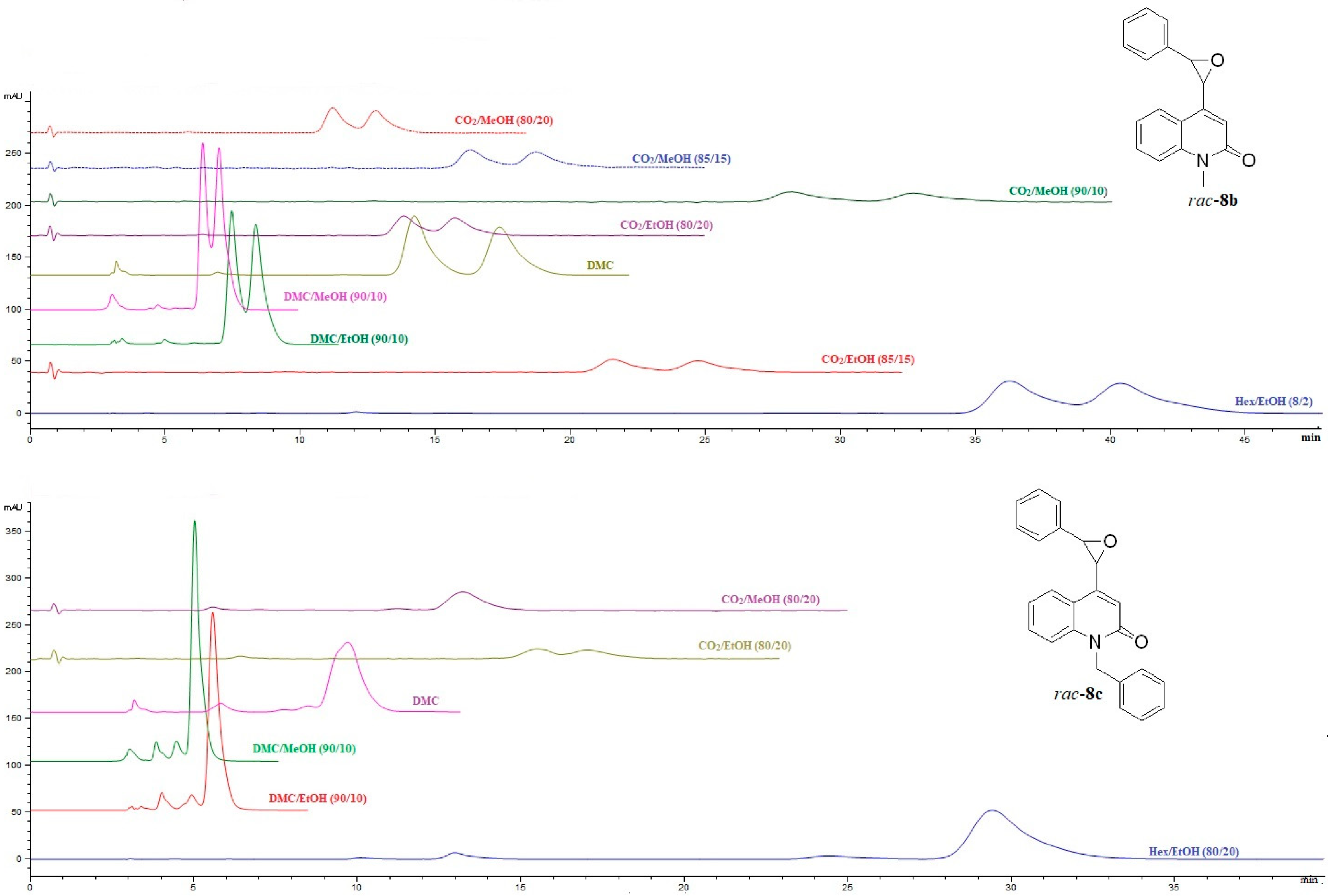
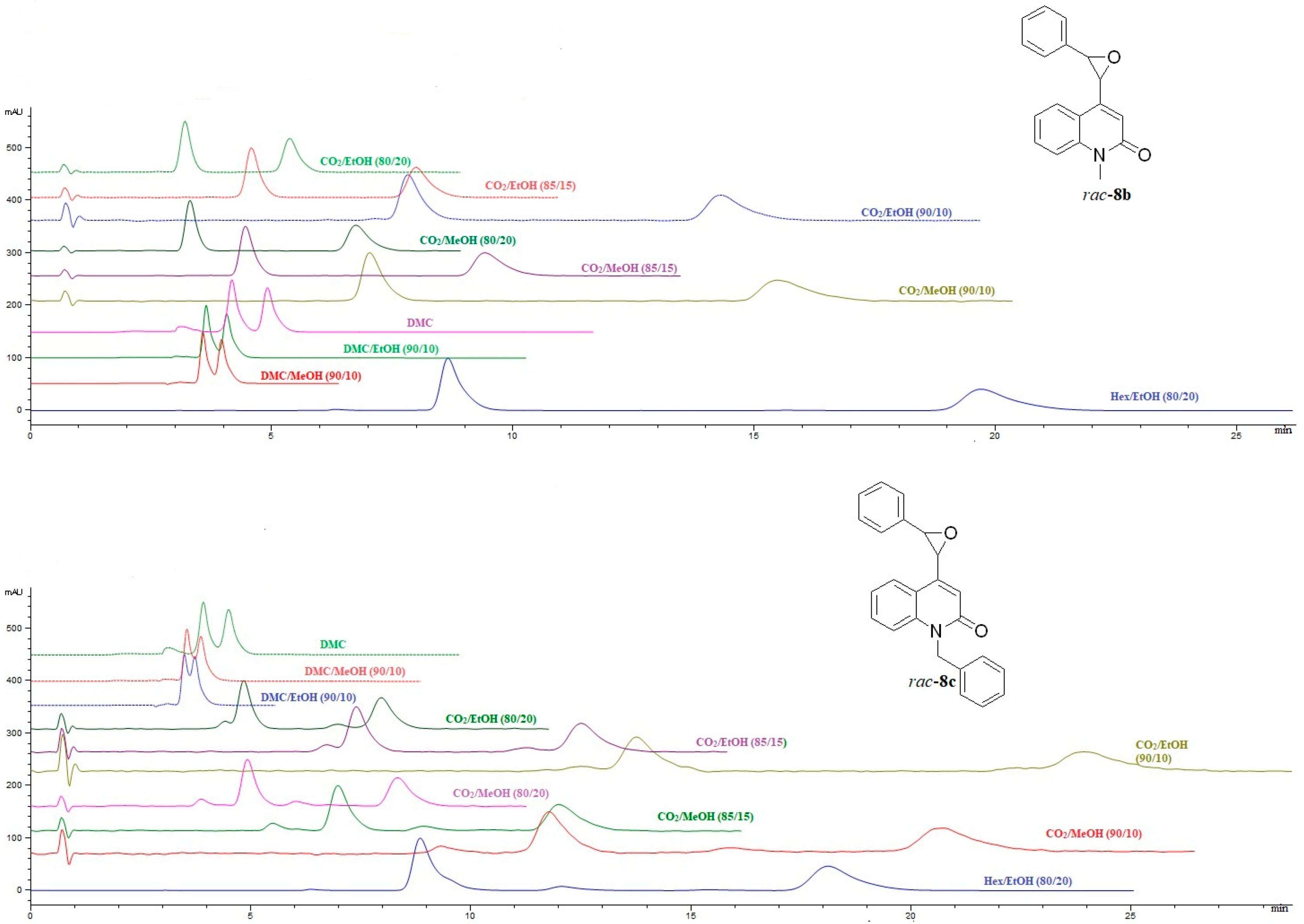
2.2.2. HPLC and SFC Enantioselective Chromatography of (±)-Disubstituted Marinoepoxides rac-8a–c
The results of enantioseparation of marinoepoxides
rac-
8a–
c on cellulose-based chiral stationary phase CHIRAL ART Cellulose-SC, and amylose-based chiral stationary phase CHIRAL ART Amylose-SA are shown in
Table S2 (Supplementary materials). It is interesting to note that the analytes
rac-
8a–
c showed a higher degree of enantioselectivity on the CHIRAL ART Amylose-SA column than on the CHIRAL ART Cellulose-SC column, regardless of the mobile phase composition (
Figure 5 and
Figure 6).
It is known from the literature that the most common interactions of the chiral selector present in the CHIRAL ART Cellulose-SC and analyte are dipole-dipole interactions and hydrogen bonds [
35]. In the case of marinoepoxides
rac-
8a–
c, dipole-dipole interactions can occur via polar C=O---C=O and C-Cl---C=O group of the chiral selector and analyte, respectively. Due to the possibility of involving at least four poles in these interactions, it has been suggested that such interactions are primarily dipolar [
38,
39]. If dipole formation does not occur due to the unfavorable position of the two groups, intimate interactions can be considered, i.e., short contact or n→π* interactions [
40]. Non-covalent intermolecular hydrogen bonding can be achieved by interaction via carbamate group of chiral selector with C=O group or with oxygen in the epoxy ring of marinoepoxides. In the case of compound
rac-
8a, there are two additional possibilities with regard to the literature that quinoline-2(1
H)-ones can occur in two tautomeric forms [
41]. If the molecule is in the enol form, then the hydrogen bond can be realized through the OH group in the analyte structure with the C=O group of the chiral selector. By applying
n-hexane/ethanol (80/20,
v/
v) mobile phase, only partial separation of the enantiomers of compounds
rac-
8a,
b was achieved, with long retention of the enantiomers of
rac-
8b (
k1 = 36.26,
k2 = 40.36) on the tested cellulose columns. By comparing the resolution factors of compounds
rac-
8a and
rac-
8b, it was observed that their
Rs values follow the sequence:
Rs (
rac-
8b) >
Rs (
rac-8a). Such results contribute to the thesis that the dipole-dipole interaction and hydrogen bond dominate in the chiral recognition mechanism. Given the structural difference between the compounds
rac-
8a–
c, it can be assumed that the dominant interactions affecting the chiral recognition mechanism are realized through the C=O group in the analyte structure. Since compound
rac-
8b showed the best enantioselectivity, regardless of the composition of the mobile phase in the HPLC mode, it can be concluded that the electron-donating methyl group has a great influence, which increases the electron density of the internal amide bond and thus contributes to stronger interactions.
Using dimethyl carbonate as the mobile phase, the best results of enantiomer separation were achieved, and the enantiomers of the two compounds rac-8a (α = 1.23; Rs = 1.49) and rac-8b (α = 1.28; Rs = 1.85) showed similar enantioselectivity. The addition of 10% methanol or 10% ethanol to the mobile phase reduces the degree of chiral recognition with the stationary phase selector in CHIRAL ART Cellulose-SC. It should also be noted that the retention times of the analyte on the column, which can be seen by observing the k1 and k2 data, are much shorter compared to the retention times of the enantiomer with dimethyl carbonate as the mobile phase. The planar carbonyl group of the marinoepoxides rac-8a–c has a relatively large dipole moment and therefore has a high solvating power. Polar molecules of methanol or ethanol surround the carbonyl group of the analyte and it becomes sterically disturbed, which may affect the weaker interactions with the chiral stationary phase selector.
The use of ethanol (
v/
v) in enantioseparation of
rac-
8a achieved better chiral recognition and slightly longer retention times of the enantiomers (
Rs = 1.24;
k1 = 2.10;
k2 = 2.49) than in comparison with methanol (
Rs = 0.69;
k1 = 0.93;
k2 = 1.08). The absorbency of the analyte on the chiral stationary phase, together with the retention factor, is increased by decreasing the volume fraction of the alcohol modifier. By reducing the polarity of the mobile phase, the interaction between the enantiomers of the analyte and the stationary phase is significantly increased, which causes longer retention times of enantiomers and their better separation [
42]. Longer retention times can consequently lead to circular and longitudinal diffusion [
28].
Better chiral recognition of compounds
rac-
8a and
rac-
8b with the chiral selector
tris-(3,5-dichlorophenylcarbamoyl)cellulose was not achieved by the use of SFC, regardless of the type and volume of alcohol modifier in the CO
2 binary system. According to the results obtained, it was observed that the best enantioselectivity was shown by compound
rac-
8b having a methyl group attached to the internal amide bond of quinoline-2(1
H)-one moiety. By comparing the retention factors of the first-emerging enantiomers of compounds
rac-
8a–
c on the selected cellulose-based chiral stationary phase, the longest retention for enantiomers of
rac-
8c having a benzyl group attached to the internal amide bond within the quinoline-2(1
H)-one structure was observed. By examining the effect of the addition of an alcohol modifier in the CO
2 mobile phase, it was found that compounds
rac-
8a-
b showed better enantioselectivity by the addition of ethanol compared to more polar methanol. Reducing the volume fraction of ethanol in the CO
2 binary system leads to lower enantioselectivity for
rac-
8a-
b. It was also observed that reducing the volume fraction of alcohol in the CO
2 binary system leads to longer retention times of the analyte, and at the same time to higher values of the resolution factor
Rs [
28]. Enantioseparation of
rac-
8c with CO
2/EtOH (85/15,
v/
v) showed slightly better enantioselectivity with a resolution factor
Rs = 1.20 and a retention factor
k1 = 25.26 of the first enantiomer, relative to CO
2/EtOH (80/20,
v/
v) as the mobile phase. It is interesting to note that with CO
2/MeOH (80/20,
v/
v) there is a complete lack of enantioseparation. A change in the polarity and volume of alcohol is sufficient to change the geometry and/or size of chiral cavities, which may affect the chiral recognition mechanism [
26,
43,
44,
45]. Polar alcohols will form stronger hydrogen bonds with the chiral stationary phase, due to the fact that they can diffuse more easily into a well-defined chiral cavity of the stationary phase [
33]. Therefore, less stable complexes will be formed, which will then lead to lower
Rs and α. Consistent with the results obtained in the HPLC mode, it was observed that the enantiomers of the analyte were retained shorter with methanol in the CO
2 binary system than with ethanol.
Comparing the results of the enantioselective analysis, it was observed that the enantiomers of compound rac-8b with a methyl group attached to the internal amide bond of quinoline-2(1H)-one showed the best chiral recognition with chiral selector tris-(3,5-dichlorophenylcarbamoyl)cellulose, both in HPLC and SFC mode. In the HPLC mode, the enantiomers of compound rac-8b are retained the longest with the best enantioselectivity, while the longest retention time in the SFC mode was observed for the enantiomers of compound rac-8c with the lower enantioselectivity. Comparing the results of both modes, it was observed that compound rac-8b showed the best enantioseparation results (α = 1.28, Rs = 1.85) with dimethyl carbonate as the mobile phase.
When the type of chiral stationary phase in the enantioselective system changes from the CHIRAL ART Cellulose-SC to the CHIRAL ART Amylose-SA, the chiral recognition mechanism also changes. In amylose, the geometry of the α-1,4-glycosidic bond between D-(+)-glucose units causes the natural bending of the associated glucopyranoses into a hollow helix. With such spatial organization, glucose units are properly distributed and make well-defined chiral cavities [
46]. A potentially stronger hydrogen bond can only be achieved in the case of compound
rac-
8a via the carbamate group of the chiral selector and hydrogen of the amide group in the analyte structure when it is in the keto form. If compound
rac-
8a is in the enol form, it is possible to form a hydrogen bond via the carbamate group of the chiral selector and the hydrogen atom of the hydroxyl group in the analyte structure. In addition to affecting the hydrogen acidity of the phenylcarbamate group, methyl groups also have a role in controlling the entry of enantiomers into the chiral cavity of the amylose chiral stationary phase [
47].
Based on the obtained results, excellent enantiorecognition of compounds
rac-
8a–
c with
tris-(3,5-dimethylphenylcarbamoyl)amylose chiral selector (CHIRAL ART Amylose SA column) can be observed using both HPLC and SFC. All marinoepoxide structures
rac-
8a–
c showed efficient chiral recognition and shorter enantiomer retention times, regardless of the mobile phase composition in HPLC and SFC mode compared to CHIRAL ART Cellulose-SC. The results of enantioseparation of compounds
rac-
8a–
c on amylose-based chiral stationary phase indicate that in the mechanism of chiral recognition, in addition to noncovalent dipole-dipole and π-π interactions, the placement of diastereomeric complex in the chiral cavity of the selector has a significant contribution. As the tertiary structure of amylose is different from cellulose, it is to be expected that the mechanism of chiral recognition will be different. The higher structure of the amylose coil in this case favors the entry of the less sterically demanding enantiomer [
43]. Using dimethyl carbonate as the mobile phase, worse enantiomer separation results are obtained for marinoepoxides
rac-
8a–
c on the amylose-based chiral stationary phase. The highest observed value of the separation factor was α = 1.63 for compound
rac-
8c, while compound
rac-
8c showed slightly less enantioselectivity with α = 1.61. Examination of the influence of different volumes of alcohol in dimethyl carbonate mobile phase on the separation of enantiomers of compound
rac-
8a–
c showed the dependence of chiral recognition on the percentage and type of alcohol modifier in the mobile phase. The addition of 10% methanol and 10% ethanol, respectively, reduces the retention factor and the resolution factor of compounds
rac-
8b–
c. The values of the separation factors for compounds
rac-
8a and
rac-
8b are higher with the addition of the alcohol modifier to the dimethyl carbonate mobile phase. Compound
rac-
8a showed slightly better enantioselectivity by the addition of methanol, with a longer retention time of the enantiomers than with ethanol. On the other hand, the enantiomers of compounds
rac-
8b and
rac-
8c showed slightly better enantiorecognition with the addition of ethanol than with the addition of methanol. The obtained results indicate that the chiral recognition of the enantiomers of compounds
rac-
8a–
c with the selector of the tested amylose-based stationary phase depends on the substituents on the nitrogen atom of the internal amide bond, the mobile phase, but also on the supramolecular structure of the stationary phase.
Better enantioselectivity was achieved in the SFC mode compared to the HPLC mode for the selected amylose-based chiral stationary phase. As shown in
Table S2 (Supplementary materials), all marinoepoxides
rac-
8a–
c were efficiently baseline separated with high selectivity and resolution and shorter retention times on the amylose-based column with CO
2 binary mobile phase. If we compare the enantiorecognition of compounds
rac-
8a–
c, then it can be concluded that the best chiral recognition using
tris-(3,5-dimethylphenylcarbamoyl) amylose selector was achieved with compound
rac-
8a having a free, non-substituted amide bond in quinoline-2(1
H)-one moiety, regardless of composition and volume fraction of alcohol modifier in the CO
2 binary system. While the lower enantiorecognition was achieved with a sterically more demanding compound
rac-
8c having a benzyl group attached to the nitrogen of the internal amide bond. These results indicated that the change in the shape of the chiral cavity is less conducive to the sterically more demanding compound
rac-
8c. By comparing the retention factors of the first eluted enantiomers of compounds
rac-
8a–
c on the selected amylose stationary phase, the longest retention of enantiomers of compound
rac-
8a with unprotected internal amide bond is observed. By examining the effect of the addition of an alcohol modifier on the CO
2 mobile phase, it was found that all compounds
rac-
8a–
c showed better enantioselectivity by the addition of methanol compared to less polar ethanol. An enantioselective chromatographic system with methanol as an alcohol modifier was more efficient for separating the enantiomers of all three compounds. By comparing separation factors and resolution factors with CO
2/MeOH (80/20,
v/
v) as the mobile phase, compound
rac-
8a showed the best enantioselectivity (α = 3.11) with the highest resolution factor (
Rs = 6.33). Binding of the methyl and benzyl groups to the nitrogen atom of the internal amide bond of compounds
rac-
8b,
c decreases the enantioselectivity. The sterically more demanding compound
rac-
8c showed the lower enantioselectivity within this group. It has also been observed that a decrease in the alcohol ratio in the binary system caused a longer retention of the enantiomers of
rac-
8a–
c on the column, with an increase in the resolution factor. The best selectivity and resolution were achieved with CO
2/ MeOH (90/10,
v/
v) for all marinoepoxides in this group. The bulky and planar benzyl substituent in compound
rac-
8c is sterically less suitable for the existing shape or size of the chiral cavity, while the less sterically demanding compound
rac-
8a is more suitable for placement in selector grooves. It should be also noted that compound
rac-
8a can achieve intensely strong hydrogen bonding via the NH group of the internal amide bond with the C=O group in the phenylcarbamate unit of the chiral selector, which may contribute to additional stabilization of the resulting diastereomeric complex or better chiral recognition.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




