
Synthesis of 6-Halo-Substituted Pericosine A and an Evaluation of Their Antitumor and Antiglycosidase Activities

 ,
,
Abstract
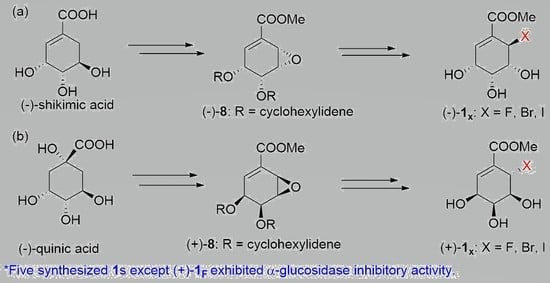
:
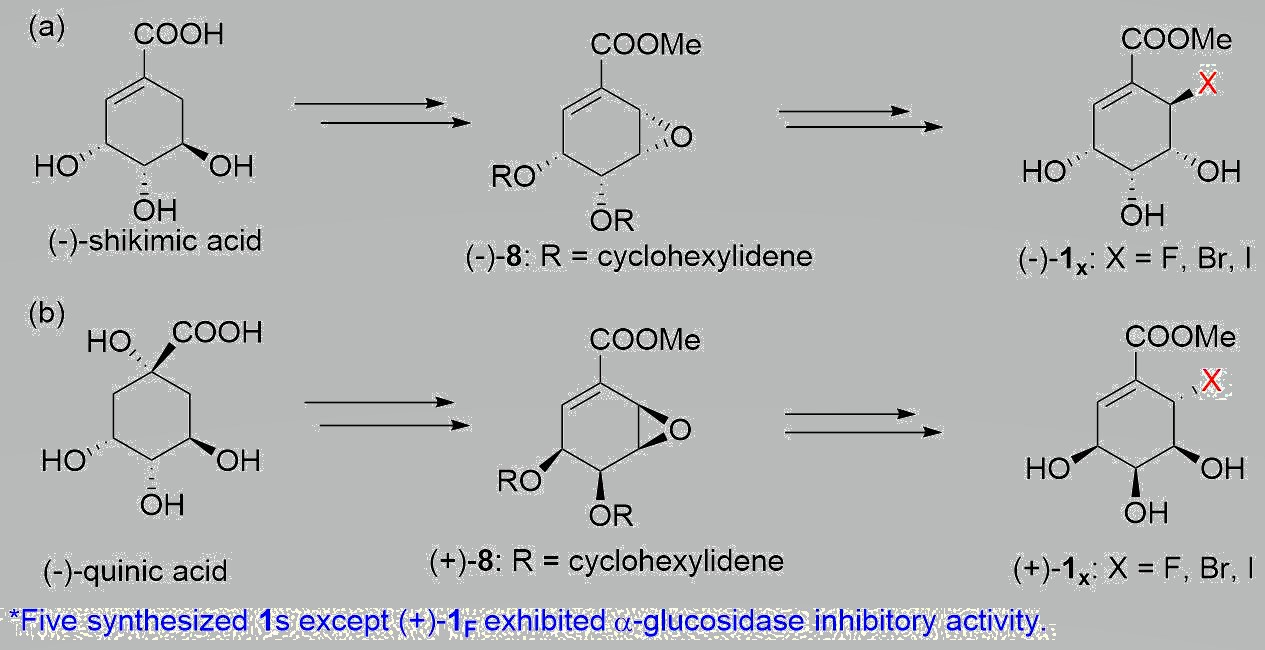
1. Introduction
2. Results & Discussion
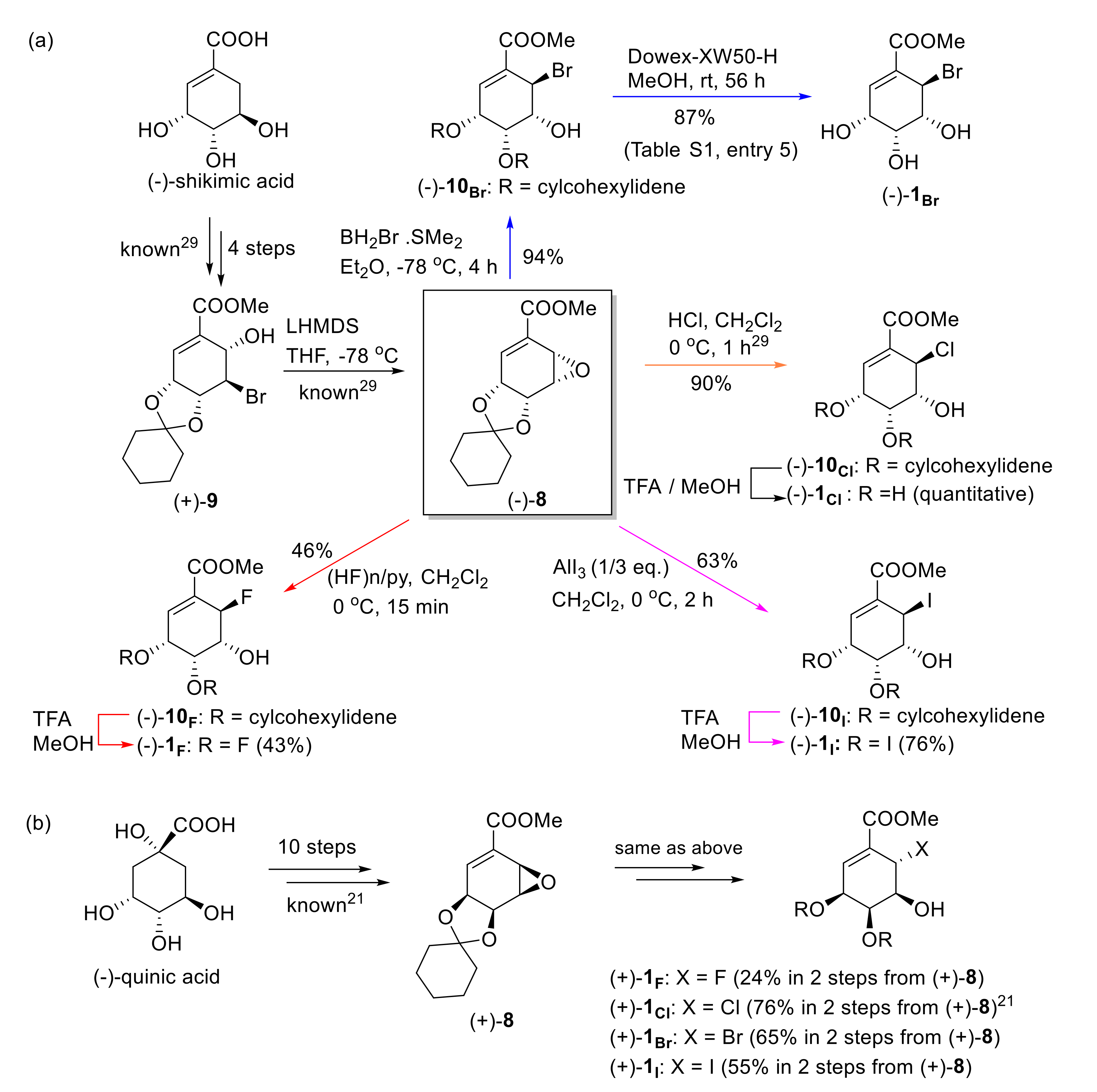
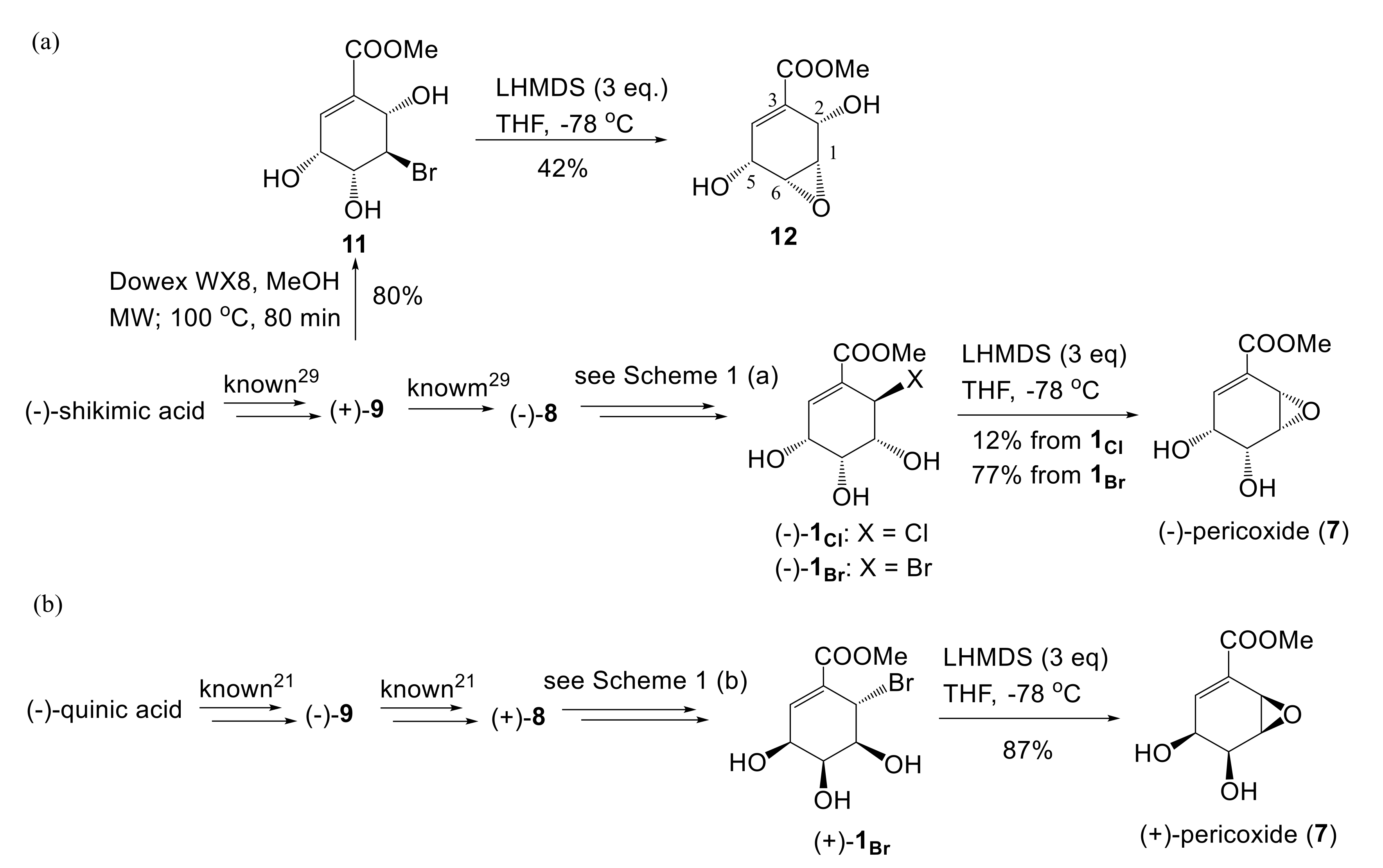
2.1. Synthesis of Both Enantiomers of 6-Halo-Substituted Pericosine A
2.2. Synthesis of Pericoxide
2.3. Evaluation of the Biological Activities of Enantiomerically Pure Pericosine A and Its 6-Halogenated Congeners
2.3.1. Antitumor Assay
2.3.2. Glycosidase Inhibitory Activity Assay
3. Materials and Methods
3.1. Synthesis of Both Enantiomers of The 6-Halopericosine A Analogs
3.1.1. Synthesis of Methyl (−)-3,4-O-Cyclohexylidene-6-Fluoro-3,4,5-Trihydroxy-1-CycloHexene Carboxylate (10F)
3.1.2. Synthesis of (−)-6-Fluoropericosine A (1F)
3.1.3. Synthesis of Methyl (−)-6-Bromo-3,4-O-Cyclohexylidene-3,4,5-Trihydroxy-1-CycloHexene Carboxylate (10Br)
3.1.4. Synthesis of (−)-6-Bromopericosine A (1Br)
3.1.5. Synthesis of Methyl (−)-3,4-O-Cyclohexylidene-3,4,5-Trihydroxy-6-iodo-1-CycloHexene Carboxylate (10I)
3.1.6. Synthesis of (−)-6-Iodoopericosine A (1I)
3.2. Synthesis of Pericoxide
Synthesis of Methyl (3R,4R,5R,6S)-5-Bromo-3,4,6-Trihydroxycyclohex-1-ene-1-carBoxylate (11) from 9
3.3. Intramolecular Epoxidation of Bromotriol 11
3.4. Synthesis of (−)-Pericoxide (7)
3.5. Biological Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gerry, C.J.; Schreiber, S.L. Chemical probes and drug leads from advances in synthetic planning and methodology. Nat. Rev. Drug Discov. 2018, 17, 333–352. [Google Scholar] [CrossRef] [PubMed]
- Campos, K.R.; Coleman, P.J.; Alvarez, J.C.; Dreher, S.D.; Garbaccio, R.M.; Terrett, N.K.; Tillyer, R.D.; Truppo, M.D.; Parmee, E.R. The importance of synthetic chemistry in the pharmaceutical industry. Science 2019, 363, eaat0805. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H. Discovery and development of natural product-derived chemotherapeutic agents based on a medicinal chemistry approach. J. Nat. Prod. 2010, 73, 500–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szychowski, J.; Truchon, J.F.; Bennani, Y.L. Natural products in medicine: Transformational outcome of synthetic chemistry. J. Med. Chem. 2014, 57, 9292–9308. [Google Scholar] [CrossRef] [PubMed]
- Ermert, P. Design, properties and recent application of macrocycles in medicinal chemistry. Chimia 2017, 71, 678–702. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.; Broenstrup, M. Industrial natural product chemistry for drug discovery and development. Nat. Prod. Rep. 2014, 31, 35–60. [Google Scholar] [CrossRef]
- Koehn, F.E. Biosynthetic medicinal chemistry of natural product drugs. Med. Chem. Comm. 2012, 3, 854–865. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M.; Shahabi, D.; Chapman, M.E.; Mesecar, A.D. Drug development and medicinal chemistry efforts toward SARS-Coronavirus and COVID-19 therapeutics. Chem. Med. Chem. 2020, 15, 907–993. [Google Scholar] [CrossRef]
- Lu, W.Y.; Li, H.J.; Li, Q.Y.; Wu, Y.C. Application of marine natural products in drug research. Bioorg. Med. Chem. 2021, 35, 11605. [Google Scholar] [CrossRef]
- Villa, F.A.; Gerwick, L. Marine natural product drug discovery: Leads for treatment of inflammation, cancer, infections, and neurological disorders. Immunopharmacol. Immunotoxicol. 2010, 32, 228–237. [Google Scholar] [CrossRef]
- Kobayashi, J. Search for new bioactive marine natural products and application to drug development. Chem. Pharm. Bull. 2016, 64, 1079–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.E.F.; Moustafa, M.S.; Abd El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine natural products: A source of novel anticancer drugs. Mar. Drugs 2019, 17, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numata, A.; Iritani, M.; Yamada, T.; Minoura, K.; Matsumura, E.; Yamori, T.; Tsuruo, T. Novel antitumour metabolites produced by a fungal strain from a sea hare. Tetrahedron Lett. 1997, 38, 8215–8218. [Google Scholar] [CrossRef]
- Yamada, T.; Iritani, M.; Ohishi, H.; Tanaka, K.; Doi, M.; Minoura, K.; Numata, A. Pericosines, antitumour metabolites from the sea hare-derived fungus Periconia byssoides. Structures and biological activities. Org. Biomol. Chem. 2007, 5, 3979–3986. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y. Synthesis of marine-derived carbasugar pericosines. In Studies in Natural Product Chemistry; Atta-ur-Rahman, A.Z., Ed.; Pergamon: Oxford, UK, 2014; Volume 41, pp. 287–319. [Google Scholar]
- Arjona, O.; Gomez, A.M.; Lopez, J.C.; Plumet, J. Synthesis, and conformational and biological aspects of carbasugars. Chem. Rev. 2007, 107, 1919–2036. [Google Scholar] [CrossRef]
- Lahiri, R.; Ansari, A.A.; Vankar, Y.D. Carbasugars and related molecules as glycosidase inhibitors. Chem. Soc. Rev. 2013, 42, 5102–5118. [Google Scholar] [CrossRef]
- Ogawa, S.; Kanto, M.; Suzuki, Y. Development and medical application of unsaturated carbaglycosylamine glycosidase inhibitors. Mini-Rev. Med. Chem. 2007, 7, 679–691. [Google Scholar] [CrossRef]
- Donohoe, T.J.; Blades, K.; Helliwell, M.; Warning, M.J.; Newcombe, N.J. The synthesis of (+)-pericosine B. Tetrahedron Lett. 1998, 39, 8755–8758. [Google Scholar] [CrossRef]
- Usami, Y.; Takaoka, I.; Ichikawa, H.; Horibe, Y.; Tomiyama, S.; Ohtsuka, M.; Imanishi, Y.; Arimoto, M. First total synthesis of antitumor natural product (+)- and (−)-pericosine A: Determination of absolute stereostructure. J. Org. Chem. 2007, 72, 6127–6134. [Google Scholar] [CrossRef]
- Usami, Y.; Ohsugi, M.; Mizuki, K.; Ichikawa, H.; Arimoto, M. Facile and efficient synthesis of naturally occurring carbasugars (+)-pericosines A and C. Org. Lett. 2009, 11, 2699–2701. [Google Scholar] [CrossRef]
- Usami, Y.; Suzuki, K.; Mizuki, K.; Ichikawa, H.; Arimoto, M. Synthesis of (−)-pericosine B, antipode of cytotoxic marine natural product. Org. Biomol. Chem. 2009, 7, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Boyd, D.R.; Sharma, N.D.; Acaru, C.A.; Malone, J.F.; O’Dowd, C.R.; Allen, C.C.R.; Stevenson, P.J. Chemoenzymatic synthesis of carbasugars (+)-pericosines A–C from diverse aromatic cis-dihydrodiol precursors. Org. Lett. 2010, 12, 2206–2209. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Shaikh, A.C.; Chen, C. Facile carbohydrate-based stereocontrolled divergent synthesis of (+)-pericosines A and B. Org. Biomol. Chem. 2011, 9, 7306–7308. [Google Scholar] [CrossRef] [PubMed]
- Reddy, Y.S.; Kadigachalam, P.; Basak, R.K.; Pal, A.P.J.; Vankar, Y.D. Total synthesis of (+)-pericosine B and (+)-pericosine C and their enantiomers by using the Baylis–Hillman reaction and ring-closing metathesis as key steps. Tetrahedron Lett. 2012, 53, 132–136. [Google Scholar] [CrossRef]
- MuniRaju, C.; Rao, J.P.; Rao, B.V. Stereoselective synthesis of (+)-pericosine B and (+)-pericosine C using ring closing metathesis approach. Tetrahedron Asymmetry 2012, 23, 86–93. [Google Scholar] [CrossRef]
- Li, L.S.; Hou, D.R. Diastereoselective vinylalumination for the synthesis of pericosine A, B and C. RSC Adv. 2014, 4, 91–97. [Google Scholar] [CrossRef]
- Babu, D.C.; Rao, C.B.; Venkastesham, K.; Selvam, J.J.P.; Venkasteswarlu, Y. Toward synthesis of carbasugars (+)-gabosine C, (+)-COTC, (+)-pericosine B, and (+)-pericosine C. Carbohydr. Res. 2014, 388, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Mizuki, K.; Iwahashi, K.; Murata, N.; Ikeda, M.; Nakai, N.; Yoneyama, H.; Harusawa, S.; Usami, Y. Synthesis of marine natural product (−)-pericosine E. Org. Lett. 2014, 16, 3760–3763. [Google Scholar] [CrossRef]
- Bidus, N.; Banachowicz, P.; Buda, S. Application of a tandem seleno-michael/aldol reaction in the total syntheses of (+)-pericosine B, (+)-pericosine C, (+)-COTC and 7-chloro-analogue of (+)-Gabosine C. Tetrahedron 2020, 76, 131397. [Google Scholar] [CrossRef]
- Usami, Y.; Mizuki, K.; Kawahata, R.; Shibano, M.; Sekine, A.; Yoneyama, H.; Harusawa, S. Synthesis of natural O-linked carba-disaccharides, (+)- and (–)-pericosine E, and their analogues as α-glucosidase inhibitors. Mar. Drugs 2017, 15, 22. [Google Scholar] [CrossRef] [Green Version]
- Usami, Y.; Higuchi, M.; Mizuki, K.; Yamamoto, M.; Kanki, M.; Nakasone, C.; Sugimoto, Y.; Shibano, M.; Uesawa, Y.; Nagai, J.; et al. Syntheses and glycosidase inhibitory activities, and in silico docking studies of pericosine E analogs methoxy-substituted at C6. Mar. Drugs 2020, 18, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Robles, A.J.; King, J.B.; Powell, D.R.; Miller, A.N.; Mooberry, S.L.; Cichewicz, R.H. Crowd sourcing natural products discovery to access uncharted dimensions of fungal metabolite diversity. Angew. Chem. Int. Ed. 2014, 53, 804–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; You, J.; Nicholas, K.M.; Cichewicz, R.H. Chemoreactive natural products that afford resistance against disparate antibiotics and toxins. Angew. Chem. Int. Ed. 2016, 55, 4220–4225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usami, Y.; Nakamura, K.; Mizobuchi, Y.; Yoneyama, H.; Harusawa, S.; Yamada, T. Enantiomeric composition of natural pericosine A derived from Periconia byssoides and α-glycosidase inhibitory activity of (–)-enantiomer. Chirality 2022, 1–8. [Google Scholar] [CrossRef]
- Brown, H.C.; Roy, C.D. Dibromoborane-dimethyl sulfide and monobromoborane–dimethyl-sulfide as superior reagents for the opening of oxiranes to bromohydrins. Molecules 1998, 2, 114–120. [Google Scholar] [CrossRef]
- Roy, C.D.; Brown, H.C. Monobromoborane-dimethyl sulfide-a highly promising reagent for the regio- and chemoselective brominative cleavage of terminal epoxides into vicinal bromohydrins. Aust. J. Chem. 2007, 60, 139–145. [Google Scholar] [CrossRef]
- Bhatt, M.V.; Babu, J.M. New reagents 3: Alumi.ium iodide—A highly regioselective ether-cleaving reagent with novel cleavage pattern. Tetrahedron Lett. 1984, 25, 3497–3500. [Google Scholar]
- Du, L.; Munteanu, C.; King, J.B.; Frantz, D.E.; Cichewicz, R.H. An electrophilic natural product provides a safe and robust odor neutralization approach to counteract malodorous organosulfur metabolites encountered in skunk spray. J. Nat. Prod. 2019, 82, 1989–1999. [Google Scholar] [CrossRef]
- Kumar, K.S.A.; Rathee, J.S.; Subramanian, M.; Chattopadhyay, S. Divergent synthesis of 4-epi-fagomine, 3,4-dihydroxypipecolic acid, and a dihydroxyindolizidine and their β-galactosidase inhibitory and immunomodulatory activities. J. Org. Chem. 2013, 78, 7406–7413. [Google Scholar] [CrossRef]
- Dada, L.; Manzano, V.E.; Varela, O. Design and synthesis of 2-acetamido-2,3-dideoxythiodisaccharides via diastereoselective conjugate addition to sugar enone O-acetyl oximes. galactosidase inhibition studies. Org. Lett. 2018, 20, 6225–6228. [Google Scholar] [CrossRef]
- Front, S.; Gallienne, E.; Charollais-Thoenig, J.; Demotz, S.; Martin, O.R. N-Alkyl-, 1-C-silyl-, and 5-C-Alkyl-1,5-dideoxy-1,5-imino-(L)-ribitols as galactosidase inhibitors. Chem. Med. Chem. 2016, 11, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Govindaraj, R.G.; Manavalan, B.; Lee, G.; Choi, S. Molecular modeling-based evaluation of hTLR10 and identification of potential ligands in toll-like receptor signaling. PLoS ONE 2010, 5, e12713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Miyake, H.; Kusunoki, M.; Osaki, S. Crystal structures of isomaltase from Saccharomyces cerevisiae and in complex with its competitive inhibitor maltose. FEBS J. 2010, 277, 4205–4214. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Zhao, D.; Xue, Z. Exploring the interaction between Salvia miltiorrhiza and α-glucosidase: Insights from computational analysis and experimental studies. RSC Adv. 2018, 8, 24701–24710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinowsky, L.; Weber, J.; Balasupramaniam, S.; Baumann, K.; Proschak, E. A diverse benchmark based on 3D matched molecular pairs for validating scoring functions. ACS Omega 2018, 3, 5704–5714. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Chavez, J.; Gonzalez-Andrade, M.; Gonzalez Mdel, C.; Glenn, A.E.; Mata, R. Thielavins A, J and K: α-Glucosidase inhibitors from MEXU 27095, an endophytic fungus from Hintonia latiflora. Phytochemistry 2013, 94, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Murugesu, S.; Ibrahim, Z.; Ahmed, Q.U.; Yusoff, N.I.N.; Uzir, B.F.; Perumal, V.; Abas, F.; Saari, K.; El-Seedi, H.; Khatib, A. Characterization of α-glucosidase inhibitors from Clinacanthus nutans lindau leaves by gas chromatography-mass spectrometry-based metabolomics and molecular docking simulation. Molecules 2018, 23, 2402. [Google Scholar] [CrossRef] [Green Version]
- Murugesu, S.; Ibrahim, Z.; Ahmed, Q.U.; Uzir, B.F.; Yusoff, N.I.N.; Perumal, V.; Abas, F.; Shaari, K.; Khatib, A. Identification of α-glucosidase inhibitors from Clinacanthus nutans leaf extract using liquid chromatography-mass spectrometry-based metabolomics and protein-ligand interaction with molecular docking. J. Pharm. Anal. 2019, 9, 91–99. [Google Scholar] [CrossRef]
- Gopalan, G.; Prabha, B.; Joe, A.; Reshmitha, T.R.; Sherin, D.R.; Abraham, B.; Sabu, M.; Manojkumar, T.K.; Radhakrishnan, K.V.; Nisha, P. Screening of Musa balbisiana Colla. seeds for antidiabetic properties and isolation of apiforol, a potential lead, with antidiabetic activity. J. Sci. Food Agric. 2019, 99, 2521–2529. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (μM) | |||
|---|---|---|---|
| Compound | P388 | L1210 | HL-60 |
| (+)-Pericosine A (1Cl)35 | 5.00 | 6.12 | 2.03 |
| (−)-Pericosine A (1Cl)35 | 4.85 | 3.96 | 2.33 |
| (+)-6-Fluoropericosine A (1F) | 9.91 | 44.0 | 10.8 |
| (−)-6-Fluoropericosine A (1F) | 9.03 | 38.0 | 9.46 |
| (+)-6-Bromopericosine A (1Br) | 5.39 | 5.66 | 5.57 |
| (−)-6-Bromopericosine A (1Br) | 5.65 | 6.30 | 6.08 |
| (+)-6-Iodopericosine A (1I) | 6.17 | 8.18 | 6.78 |
| (−)-6-Iodopericosine A (1I) | 5.91 | 8.27 | 6.45 |
| 5-FU (positive control) | 3.86 | 0.63 | 0.22 |
| IC50 (mM) | |||||
|---|---|---|---|---|---|
| Compound | α-Glucosidase a | β-Glucosidase b | α-Mannosidase c | α-Galactosidase d | β-Galactosidase e |
| (+)-1Cl35 | NI f | NI | NI | NI | NI |
| (−)-1Cl35 | 2.25 | NI | NI | NI | 5.38 |
| (+)-1F | NI | NI | NI | NI | NI |
| (−)-1F | 1.95 | NI | NI | NI | NI |
| (+)-1Br | 5.05 | NI | NI | NI | NI |
| (−)-1Br | 1.79 | NI | NI | NI | 5.60 |
| (+)-1I | 1.15 | NI | NI | 3.56 | NI |
| (−)-1I | 3.60 | NI | NI | NI | NI |
| DNJ (positive control) | 0.0965 | 0.195 | – | – | – |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usami, Y.; Mizobuchi, Y.; Ijuin, M.; Yamada, T.; Morita, M.; Mizuki, K.; Yoneyama, H.; Harusawa, S. Synthesis of 6-Halo-Substituted Pericosine A and an Evaluation of Their Antitumor and Antiglycosidase Activities. Mar. Drugs 2022, 20, 438. https://doi.org/10.3390/md20070438
Usami Y, Mizobuchi Y, Ijuin M, Yamada T, Morita M, Mizuki K, Yoneyama H, Harusawa S. Synthesis of 6-Halo-Substituted Pericosine A and an Evaluation of Their Antitumor and Antiglycosidase Activities. Marine Drugs. 2022; 20(7):438. https://doi.org/10.3390/md20070438
Chicago/Turabian StyleUsami, Yoshihide, Yoshino Mizobuchi, Mai Ijuin, Takeshi Yamada, Mizuki Morita, Koji Mizuki, Hiroki Yoneyama, and Shinya Harusawa. 2022. "Synthesis of 6-Halo-Substituted Pericosine A and an Evaluation of Their Antitumor and Antiglycosidase Activities" Marine Drugs 20, no. 7: 438. https://doi.org/10.3390/md20070438