
Targeted Large-Scale Genome Mining and Candidate Prioritization for Natural Product Discovery


Abstract
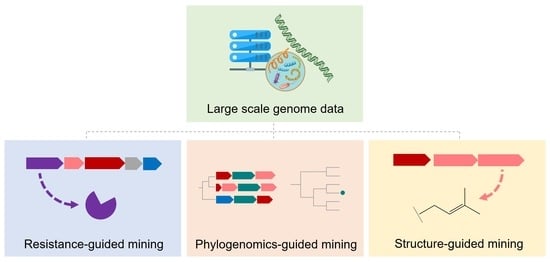
:
1. Introduction
2. Genomics for Natural Product Discovery
3. Resistance-Gene-Guided Genome Mining
4. Phylogenomics-Guided Genome Mining
5. Structure-Guided Genome Mining
6. Global Genome Mining for RiPPs
7. Conclusions
8. Future Perspective
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waglechner, N.; McArthur, A.G.; Wright, G.D. Phylogenetic Reconciliation Reveals the Natural History of Glycopeptide Antibiotic Biosynthesis and Resistance. Nat. Microbiol. 2019, 4, 1862–1871. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; Wang, Y.; Lui Ang, E.; Zhao, H. Engineering Microbial Hosts for Production of Bacterial Natural Products. Nat. Prod. Rep. 2016, 33, 963–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hug, J.J.; Krug, D.; Müller, R. Bacteria as Genetically Programmable Producers of Bioactive Natural Products. Nat. Rev. Chem. 2020, 4, 172–193. [Google Scholar] [CrossRef]
- Breitling, R.; Ceniceros, A.; Jankevics, A.; Takano, E. Metabolomics for Secondary Metabolite Research. Metabolites 2013, 3, 1076–1083. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Kautsar, S.A.; Blin, K.; Shaw, S.; Navarro-Muñoz, J.C.; Terlouw, B.R.; van der Hooft, J.J.J.; van Santen, J.A.; Tracanna, V.; Suarez Duran, H.G.; Pascal Andreu, V.; et al. MIBiG 2.0: A Repository for Biosynthetic Gene Clusters of Known Function. Nucleic Acids Res. 2020, 48, D454–D458. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, E.C.; Schorn, M.; Larson, C.B.; Millán-Aguiñaga, N. Targeted Antibiotic Discovery through Biosynthesis-Associated Resistance Determinants: Target Directed Genome Mining. Crit. Rev. Microbiol. 2019, 45, 255–277. [Google Scholar] [CrossRef]
- Schmidt, E.W. Trading Molecules and Tracking Targets in Symbiotic Interactions. Nat. Chem. Biol. 2008, 4, 466–473. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.T.; Tang, Y. Natural Product Biosynthesis; Royal Society of Chemistry: London, UK, 2017; ISBN 978-1-78801-131-0. [Google Scholar]
- Katz, L.; Baltz, R.H. Natural Product Discovery: Past, Present, and Future. J. Ind. Microbiol. Biotechnol. 2016, 43, 155–176. [Google Scholar] [CrossRef]
- Luo, Y.; Cobb, R.E.; Zhao, H. Recent Advances in Natural Product Discovery. Curr. Opin. Biotechnol. 2014, 30, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Genilloud, O.; González, I.; Salazar, O.; Martín, J.; Tormo, J.R.; Vicente, F. Current Approaches to Exploit Actinomycetes as a Source of Novel Natural Products. J. Ind. Microbiol. Biotechnol. 2011, 38, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Wohlleben, W.; Mast, Y.; Stegmann, E.; Ziemert, N. Antibiotic Drug Discovery. Microb. Biotechnol. 2016, 9, 541–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reen, F.J.; Romano, S.; Dobson, A.D.W.; O’Gara, F. The Sound of Silence: Activating Silent Biosynthetic Gene Clusters in Marine Microorganisms. Mar. Drugs 2015, 13, 4754–4783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, B.O.; Van Lanen, S.G.; Baltz, R.H. Microbial Genome Mining for Accelerated Natural Products Discovery: Is a Renaissance in the Making? J. Ind. Microbiol. Biotechnol. 2014, 41, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Ziemert, N.; Alanjary, M.; Weber, T. The Evolution of Genome Mining in Microbes—A Review. Nat. Prod. Rep. 2016, 33, 988–1005. [Google Scholar] [CrossRef] [Green Version]
- Weber, T.; Welzel, K.; Pelzer, S.; Vente, A.; Wohlleben, W. Exploiting the Genetic Potential of Polyketide Producing Streptomycetes. J. Biotechnol. 2003, 106, 221–232. [Google Scholar] [CrossRef]
- Lee, N.; Hwang, S.; Kim, J.; Cho, S.; Palsson, B.; Cho, B.-K. Mini Review: Genome Mining Approaches for the Identification of Secondary Metabolite Biosynthetic Gene Clusters in Streptomyces. Comput. Struct. Biotechnol. J. 2020, 18, 1548–1556. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER Web Server: Interactive Sequence Similarity Searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [Green Version]
- Zerikly, M.; Challis, G.L. Strategies for the Discovery of New Natural Products by Genome Mining. ChemBioChem 2009, 10, 625–633. [Google Scholar] [CrossRef]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. AntiSMASH: Rapid Identification, Annotation and Analysis of Secondary Metabolite Biosynthesis Gene Clusters in Bacterial and Fungal Genome Sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. AntiSMASH 6.0: Improving Cluster Detection and Comparison Capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Müller, R.; Wohlleben, W.; et al. AntiSMASH 3.0—a Comprehensive Resource for the Genome Mining of Biosynthetic Gene Clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skinnider, M.A.; Johnston, C.W.; Gunabalasingam, M.; Merwin, N.J.; Kieliszek, A.M.; MacLellan, R.J.; Li, H.; Ranieri, M.R.M.; Webster, A.L.H.; Cao, M.P.T.; et al. Comprehensive Prediction of Secondary Metabolite Structure and Biological Activity from Microbial Genome Sequences. Nat. Commun. 2020, 11, 6058. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Camacho, F.R.; Wang, S.; Chankhamjon, P.; Odabas, A.; Biswas, A.; Jeffrey, P.D.; Donia, M.S. A Metagenomic Strategy for Harnessing the Chemical Repertoire of the Human Microbiome. Science 2019, 366, eaax9176. [Google Scholar] [CrossRef]
- Weber, T.; Kim, H.U. The Secondary Metabolite Bioinformatics Portal: Computational Tools to Facilitate Synthetic Biology of Secondary Metabolite Production. Synth. Syst. Biotechnol. 2016, 1, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Hadjithomas, M.; Chen, I.-M.A.; Chu, K.; Huang, J.; Ratner, A.; Palaniappan, K.; Andersen, E.; Markowitz, V.; Kyrpides, N.C.; Ivanova, N.N. IMG-ABC: New Features for Bacterial Secondary Metabolism Analysis and Targeted Biosynthetic Gene Cluster Discovery in Thousands of Microbial Genomes. Nucleic Acids Res. 2017, 45, D560–D565. [Google Scholar] [CrossRef]
- Chevrette, M.G.; Gavrilidou, A.; Mantri, S.; Selem-Mojica, N.; Ziemert, N.; Barona-Gómez, F. The Confluence of Big Data and Evolutionary Genome Mining for the Discovery of Natural Products. Nat. Prod. Rep. 2021, 38, 2024–2040. [Google Scholar] [CrossRef]
- Gerlt, J.A.; Bouvier, J.T.; Davidson, D.B.; Imker, H.J.; Sadkhin, B.; Slater, D.R.; Whalen, K.L. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A Web Tool for Generating Protein Sequence Similarity Networks. Biochim. Biophys. Acta-Proteins Proteom. 2015, 1854, 1019–1037. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Machado, H.; Tuttle, R.N.; Jensen, P.R. Omics-Based Natural Product Discovery and the Lexicon of Genome Mining. Curr. Opin. Microbiol. 2017, 39, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazzotto, E.; Weber, T. Omics and Multi-Omics Approaches to Study the Biosynthesis of Secondary Metabolites in Microorganisms. Curr. Opin. Microbiol. 2018, 45, 109–116. [Google Scholar] [CrossRef]
- Kloosterman, A.M.; Medema, M.H.; van Wezel, G.P. Omics-Based Strategies to Discover Novel Classes of RiPP Natural Products. Curr. Opin. Biotechnol. 2021, 69, 60–67. [Google Scholar] [CrossRef]
- Prihoda, D.; Maritz, J.M.; Klempir, O.; Dzamba, D.; Woelk, C.H.; Hazuda, D.J.; Bitton, D.A.; Hannigan, G.D. The Application Potential of Machine Learning and Genomics for Understanding Natural Product Diversity, Chemistry, and Therapeutic Translatability. Nat. Prod. Rep. 2021, 38, 1100–1108. [Google Scholar] [CrossRef]
- Zhong, Z.; He, B.; Li, J.; Li, Y.-X. Challenges and Advances in Genome Mining of Ribosomally Synthesized and Post-Translationally Modified Peptides (RiPPs). Synth. Syst. Biotechnol. 2020, 5, 155–172. [Google Scholar] [CrossRef]
- Hug, J.J.; Panter, F.; Krug, D.; Müller, R. Genome Mining Reveals Uncommon Alkylpyrones as Type III PKS Products from Myxobacteria. J. Ind. Microbiol. Biotechnol. 2019, 46, 319–334. [Google Scholar] [CrossRef]
- Panter, F.; Krug, D.; Baumann, S.; Müller, R. Self-Resistance Guided Genome Mining Uncovers New Topoisomerase Inhibitors from Myxobacteria. Chem. Sci. 2018, 9, 4898–4908. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Liu, Q.; Zang, X.; Yuan, S.; Bat-Erdene, U.; Nguyen, C.; Gan, J.; Zhou, J.; Jacobsen, S.E.; Tang, Y. Resistance-Gene-Directed Discovery of a Natural-Product Herbicide with a New Mode of Action. Nature 2018, 559, 415–418. [Google Scholar] [CrossRef]
- Girard, L.; Geudens, N.; Pauwels, B.; Höfte, M.; Martins, J.C.; De Mot, R. Transporter Gene-Mediated Typing for Detection and Genome Mining of Lipopeptide-Producing Pseudomonas. Appl. Environ. Microbiol. 2022, 88, e01869-21. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Abramyan, E.D.; Cheng, W.; Perlatti, B.; Harvey, C.J.B.; Bills, G.F.; Tang, Y. Targeted Genome Mining Reveals the Biosynthetic Gene Clusters of Natural Product CYP51 Inhibitors. J. Am. Chem. Soc. 2021, 143, 6043–6047. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Li, J.; Millán-Aguiñaga, N.; Zhang, J.J.; O’Neill, E.C.; Ugalde, J.A.; Jensen, P.R.; Mantovani, S.M.; Moore, B.S. Identification of Thiotetronic Acid Antibiotic Biosynthetic Pathways by Target-Directed Genome Mining. ACS Chem. Biol. 2015, 10, 2841–2849. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-X.; Zhong, Z.; Hou, P.; Zhang, W.-P.; Qian, P.-Y. Resistance to Nonribosomal Peptide Antibiotics Mediated by d-Stereospecific Peptidases. Nat. Chem. Biol. 2018, 14, 381–387. [Google Scholar] [CrossRef]
- Cimermancic, P.; Medema, M.H.; Claesen, J.; Kurita, K.; Wieland Brown, L.C.; Mavrommatis, K.; Pati, A.; Godfrey, P.A.; Koehrsen, M.; Clardy, J.; et al. Insights into Secondary Metabolism from a Global Analysis of Prokaryotic Biosynthetic Gene Clusters. Cell 2014, 158, 412–421. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Morales, P.; Kopp, J.F.; Martínez-Guerrero, C.; Yáñez-Guerra, L.A.; Selem-Mojica, N.; Ramos-Aboites, H.; Feldmann, J.; Barona-Gómez, F. Phylogenomic Analysis of Natural Products Biosynthetic Gene Clusters Allows Discovery of Arseno-Organic Metabolites in Model Streptomycetes. Genome Biol. Evol. 2016, 8, 1906–1916. [Google Scholar] [CrossRef] [Green Version]
- Culp, E.J.; Waglechner, N.; Wang, W.; Fiebig-Comyn, A.A.; Hsu, Y.-P.; Koteva, K.; Sychantha, D.; Coombes, B.K.; Van Nieuwenhze, M.S.; Brun, Y.V.; et al. Evolution-Guided Discovery of Antibiotics That Inhibit Peptidoglycan Remodelling. Nature 2020, 578, 582–587. [Google Scholar] [CrossRef]
- Ahmed, M.N.; Reyna-González, E.; Schmid, B.; Wiebach, V.; Süssmuth, R.D.; Dittmann, E.; Fewer, D.P. Phylogenomic Analysis of the Microviridin Biosynthetic Pathway Coupled with Targeted Chemo-Enzymatic Synthesis Yields Potent Protease Inhibitors. ACS Chem. Biol. 2017, 12, 1538–1546. [Google Scholar] [CrossRef]
- Mullins, A.J.; Murray, J.A.H.; Bull, M.J.; Jenner, M.; Jones, C.; Webster, G.; Green, A.E.; Neill, D.R.; Connor, T.R.; Parkhill, J.; et al. Genome Mining Identifies Cepacin as a Plant-Protective Metabolite of the Biopesticidal Bacterium Burkholderia Ambifaria. Nat. Microbiol. 2019, 4, 996–1005. [Google Scholar] [CrossRef]
- Navarro-Muñoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; De Los Santos, E.L.C.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A Computational Framework to Explore Large-Scale Biosynthetic Diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef]
- Yamada, Y.; Kuzuyama, T.; Komatsu, M.; Shin-ya, K.; Omura, S.; Cane, D.E.; Ikeda, H. Terpene Synthases Are Widely Distributed in Bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 857–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhang, C.; Zhang, L. Investigation of the Molecular Landscape of Bacterial Aromatic Polyketides by Global Analysis of Type II Polyketide Synthases. Angew. Chem. Int. Ed. 2022, 61, e202202286. [Google Scholar] [CrossRef]
- Li, Y.-X.; Zhong, Z.; Zhang, W.-P.; Qian, P.-Y. Discovery of Cationic Nonribosomal Peptides as Gram-Negative Antibiotics through Global Genome Mining. Nat. Commun. 2018, 9, 3273. [Google Scholar] [CrossRef] [PubMed]
- Malit, J.J.L.; Liu, W.; Cheng, A.; Saha, S.; Liu, L.-L.; Qian, P.-Y. Global Genome Mining Reveals a Cytochrome P450-Catalyzed Cyclization of Crownlike Cyclodipeptides with Neuroprotective Activity. Org. Lett. 2021, 23, 6601–6605. [Google Scholar] [CrossRef]
- Malit, J.J.L.; Wu, C.; Tian, X.; Liu, W.; Huang, D.; Sung, H.H.-Y.; Liu, L.-L.; Williams, I.D.; Qian, P.-Y. Griseocazines: Neuroprotective Multiprenylated Cyclodipeptides Identified through Targeted Genome Mining. Org. Lett. 2022, 24, 2967–2972. [Google Scholar] [CrossRef]
- Hudson, G.A.; Burkhart, B.J.; DiCaprio, A.J.; Schwalen, C.J.; Kille, B.; Pogorelov, T.V.; Mitchell, D.A. Bioinformatic Mapping of Radical S-Adenosylmethionine-Dependent Ribosomally Synthesized and Post-Translationally Modified Peptides Identifies New Cα, Cβ, and Cγ-Linked Thioether-Containing Peptides. J. Am. Chem. Soc. 2019, 141, 8228–8238. [Google Scholar] [CrossRef]
- Pan, G.; Xu, Z.; Guo, Z.; Hindra; Ma, M.; Yang, D.; Zhou, H.; Gansemans, Y.; Zhu, X.; Huang, Y.; et al. Discovery of the Leinamycin Family of Natural Products by Mining Actinobacterial Genomes. Proc. Natl. Acad. Sci. USA 2017, 114, E11131–E11140. [Google Scholar] [CrossRef] [Green Version]
- Ju, K.-S.; Gao, J.; Doroghazi, J.R.; Wang, K.-K.A.; Thibodeaux, C.J.; Li, S.; Metzger, E.; Fudala, J.; Su, J.; Zhang, J.K.; et al. Discovery of Phosphonic Acid Natural Products by Mining the Genomes of 10,000 Actinomycetes. Proc. Natl. Acad. Sci. USA 2015, 112, 12175–12180. [Google Scholar] [CrossRef] [Green Version]
- Walker, M.C.; Eslami, S.M.; Hetrick, K.J.; Ackenhusen, S.E.; Mitchell, D.A.; van der Donk, W.A. Precursor Peptide-Targeted Mining of More than One Hundred Thousand Genomes Expands the Lanthipeptide Natural Product Family. BMC Genom. 2020, 21, 387. [Google Scholar] [CrossRef]
- Purushothaman, M.; Sarkar, S.; Morita, M.; Gugger, M.; Schmidt, E.W.; Morinaka, B.I. Genome-Mining-Based Discovery of the Cyclic Peptide Tolypamide and TolF, a Ser/Thr Forward O-Prenyltransferase. Angew. Chem. Int. Ed. 2021, 60, 8460–8465. [Google Scholar] [CrossRef]
- Skinnider, M.A.; Johnston, C.W.; Edgar, R.E.; Dejong, C.A.; Merwin, N.J.; Rees, P.N.; Magarvey, N.A. Genomic Charting of Ribosomally Synthesized Natural Product Chemical Space Facilitates Targeted Mining. Proc. Natl. Acad. Sci. USA 2016, 113, E6343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwalen, C.J.; Hudson, G.A.; Kille, B.; Mitchell, D.A. Bioinformatic Expansion and Discovery of Thiopeptide Antibiotics. J. Am. Chem. Soc. 2018, 140, 9494–9501. [Google Scholar] [CrossRef] [PubMed]
- Santos-Aberturas, J.; Chandra, G.; Frattaruolo, L.; Lacret, R.; Pham, T.H.; Vior, N.M.; Eyles, T.H.; Truman, A.W. Uncovering the Unexplored Diversity of Thioamidated Ribosomal Peptides in Actinobacteria Using the RiPPER Genome Mining Tool. Nucleic Acids Res. 2019, 47, 4624–4637. [Google Scholar] [CrossRef]
- Bushin, L.B.; Covington, B.C.; Rued, B.E.; Federle, M.J.; Seyedsayamdost, M.R. Discovery and Biosynthesis of Streptosactin, a Sactipeptide with an Alternative Topology Encoded by Commensal Bacteria in the Human Microbiome. J. Am. Chem. Soc. 2020, 142, 16265–16275. [Google Scholar] [CrossRef] [PubMed]
- Van Heel, A.J.; Kloosterman, T.G.; Montalban-Lopez, M.; Deng, J.; Plat, A.; Baudu, B.; Hendriks, D.; Moll, G.N.; Kuipers, O.P. Discovery, Production and Modification of Five Novel Lantibiotics Using the Promiscuous Nisin Modification Machinery. ACS Synth. Biol. 2016, 5, 1146–1154. [Google Scholar] [CrossRef]
- Kling, A.; Lukat, P.; Almeida, D.V.; Bauer, A.; Fontaine, E.; Sordello, S.; Zaburannyi, N.; Herrmann, J.; Wenzel, S.C.; König, C.; et al. Targeting DnaN for Tuberculosis Therapy Using Novel Griselimycins. Science 2015, 348, 1106–1112. [Google Scholar] [CrossRef]
- Kale, A.J.; McGlinchey, R.P.; Lechner, A.; Moore, B.S. Bacterial Self-Resistance to the Natural Proteasome Inhibitor Salinosporamide A. ACS Chem. Biol. 2011, 6, 1257–1264. [Google Scholar] [CrossRef] [Green Version]
- Peterson, R.M.; Huang, T.; Rudolf, J.D.; Smanski, M.J.; Shen, B. Mechanisms of Self-Resistance in the Platensimycin and Platencin Producing Streptomyces Platensis MA7327 and MA7339 Strains. Chem. Biol. 2014, 21, 389–397. [Google Scholar] [CrossRef] [Green Version]
- Amorim Franco, T.M.; Blanchard, J.S. Bacterial Branched-Chain Amino Acid Biosynthesis: Structures, Mechanisms, and Drugability. Biochemistry 2017, 56, 5849–5865. [Google Scholar] [CrossRef] [Green Version]
- Sélem-Mojica, N.; Aguilar, C.; Gutiérrez-García, K.; Martínez-Guerrero, C.E.; Barona-Gómez, F. EvoMining Reveals the Origin and Fate of Natural Product Biosynthetic Enzymes. Microb. Genom. 2019, 5, e000260. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic Resistome Surveillance with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.K.; Forsberg, K.J.; Dantas, G. Improved Annotation of Antibiotic Resistance Determinants Reveals Microbial Resistomes Cluster by Ecology. ISME J. 2015, 9, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Fischbach, M.A. Computational Approaches to Natural Product Discovery. Nat. Chem. Biol. 2015, 11, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Baltz, R.H. Natural Product Drug Discovery in the Genomic Era: Realities, Conjectures, Misconceptions, and Opportunities. J. Ind. Microbiol. Biotechnol. 2019, 46, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Baltz, R.H. Genome Mining for Drug Discovery: Progress at the Front End. J. Ind. Microbiol. Biotechnol. 2021, 48, kuab044. [Google Scholar] [CrossRef]
- Palaniappan, K.; Chen, I.-M.A.; Chu, K.; Ratner, A.; Seshadri, R.; Kyrpides, N.C.; Ivanova, N.N.; Mouncey, N.J. IMG-ABC v.5.0: An Update to the IMG/Atlas of Biosynthetic Gene Clusters Knowledgebase. Nucleic Acids Res. 2020, 48, D422–D430. [Google Scholar] [CrossRef]
- Komatsu, M.; Tsuda, M.; Ōmura, S.; Oikawa, H.; Ikeda, H. Identification and Functional Analysis of Genes Controlling Biosynthesis of 2-Methylisoborneol. Proc. Natl. Acad. Sci. USA 2008, 105, 7422–7427. [Google Scholar] [CrossRef] [Green Version]
- Harken, L.; Li, S.-M. Modifications of Diketopiperazines Assembled by Cyclodipeptide Synthases with Cytochrome P450 Enzymes. Appl. Microbiol. Biotechnol. 2021, 105, 2277–2285. [Google Scholar] [CrossRef]
- Haft, D.H.; Basu, M.K. Biological Systems Discovery in Silico: Radical S-Adenosylmethionine Protein Families and Their Target Peptides for Posttranslational Modification. J. Bacteriol. 2011, 193, 2745–2755. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wang, J.; Li, G.; Yang, Y.; Ding, W. Current Advancements in Sactipeptide Natural Products. Front. Chem. 2021, 9, 595991. [Google Scholar] [CrossRef]
- Montalbán-López, M.; Scott, T.A.; Ramesh, S.; Rahman, I.R.; van Heel, A.J.; Viel, J.H.; Bandarian, V.; Dittmann, E.; Genilloud, O.; Goto, Y.; et al. New Developments in RiPP Discovery, Enzymology and Engineering. Nat. Prod. Rep. 2021, 38, 130–239. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.T.Y.; Gellatly, S.L.; Hancock, R.E.W. Multifunctional Cationic Host Defence Peptides and Their Clinical Applications. Cell. Mol. Life Sci. 2011, 68, 2161. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Vogel, H.J. Diversity of Antimicrobial Peptides and Their Mechanisms of Action. Biochim. Biophys. Acta-Biomembr. 1999, 1462, 11–28. [Google Scholar] [CrossRef] [Green Version]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products: Overview and Recommendations for a Universal Nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, A.M.; Shelton, K.E.; van Wezel, G.P.; Medema, M.H.; Mitchell, D.A. RRE-Finder: A Genome-Mining Tool for Class-Independent RiPP Discovery. mSystems 2020, 5, 267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, M.; Bruner, S.D.; Ding, Y. Heterologous Production of Microbial Ribosomally Synthesized and Post-Translationally Modified Peptides. Front. Microbiol. 2018, 9, 1801. [Google Scholar] [CrossRef]
- Malit, J.J.L.; Wu, C.; Liu, L.-L.; Qian, P.-Y. Global Genome Mining Reveals the Distribution of Diverse Thioamidated RiPP Biosynthesis Gene Clusters. Front. Microbiol. 2021, 12, 987. [Google Scholar] [CrossRef]
- Tietz, J.I.; Schwalen, C.J.; Patel, P.S.; Maxson, T.; Blair, P.M.; Tai, H.-C.; Zakai, U.I.; Mitchell, D.A. A New Genome-Mining Tool Redefines the Lasso Peptide Biosynthetic Landscape. Nat. Chem. Biol. 2017, 13, 470–478. [Google Scholar] [CrossRef]
- Singh, M.; Chaudhary, S.; Sareen, D. Roseocin, a Novel Two-Component Lantibiotic from an Actinomycete. Mol. Microbiol. 2020, 113, 326–337. [Google Scholar] [CrossRef]
- Mo, T.; Ji, X.; Yuan, W.; Mandalapu, D.; Wang, F.; Zhong, Y.; Li, F.; Chen, Q.; Ding, W.; Deng, Z.; et al. Thuricin Z: A Narrow-Spectrum Sactibiotic That Targets the Cell Membrane. Angew. Chem. Int. Ed. 2021, 58, 18793–18797. [Google Scholar] [CrossRef]
- Lee, H.; Choi, M.; Park, J.-U.; Roh, H.; Kim, S. Genome Mining Reveals High Topological Diversity of ω-Ester-Containing Peptides and Divergent Evolution of ATP-Grasp Macrocyclases. J. Am. Chem. Soc. 2020, 142, 3013–3023. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.D.; Mahanta, N.; Mitchell, D.A.; Metcalf, W.W. Post-Translational Thioamidation of Methyl-Coenzyme M Reductase, a Key Enzyme in Methanogenic and Methanotrophic Archaea. Elife 2017, 6, e29218. [Google Scholar] [CrossRef] [PubMed]
- Izawa, M.; Kawasaki, T.; Hayakawa, Y. Cloning and Heterologous Expression of the Thioviridamide Biosynthesis Gene Cluster from Streptomyces Olivoviridis. Appl. Environ. Microbiol. 2013, 79, 7110–7113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, T.; Izumikawa, M.; Kozone, I.; Hashimoto, J.; Kagaya, N.; Koiwai, H.; Komatsu, M.; Fujie, M.; Sato, N.; Ikeda, H.; et al. Neothioviridamide, a Polythioamide Compound Produced by Heterologous Expression of a Streptomyces Sp. Cryptic RiPP Biosynthetic Gene Cluster. J. Nat. Prod. 2018, 81, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Kjaerulff, L.; Sikandar, A.; Zaburannyi, N.; Adam, S.; Herrmann, J.; Koehnke, J.; Müller, R. Thioholgamides: Thioamide-Containing Cytotoxic RiPP Natural Products. ACS Chem. Biol. 2017, 12, 2837–2841. [Google Scholar] [CrossRef]
- Bushin, L.B.; Clark, K.A.; Pelczer, I.; Seyedsayamdost, M.R. Charting an Unexplored Streptococcal Biosynthetic Landscape Reveals a Unique Peptide Cyclization Motif. J. Am. Chem. Soc. 2018, 140, 17674–17684. [Google Scholar] [CrossRef]
- Caruso, A.; Bushin, L.B.; Clark, K.A.; Martinie, R.J.; Seyedsayamdost, M.R. Radical Approach to Enzymatic β-Thioether Bond Formation. J. Am. Chem. Soc. 2019, 141, 990–997. [Google Scholar] [CrossRef]
- Clark, K.A.; Bushin, L.B.; Seyedsayamdost, M.R. Aliphatic Ether Bond Formation Expands the Scope of Radical SAM Enzymes in Natural Product Biosynthesis. J. Am. Chem. Soc. 2019, 141, 10610–10615. [Google Scholar] [CrossRef]
- Caruso, A.; Martinie, R.J.; Bushin, L.B.; Seyedsayamdost, M.R. Macrocyclization via an Arginine-Tyrosine Crosslink Broadens the Reaction Scope of Radical S-Adenosylmethionine Enzymes. J. Am. Chem. Soc. 2019, 141, 16610–16614. [Google Scholar] [CrossRef]
- Schramma, K.R.; Seyedsayamdost, M.R. Lysine-Tryptophan-Crosslinked Peptides Produced by Radical SAM Enzymes in Pathogenic Streptococci. ACS Chem. Biol. 2017, 12, 922–927. [Google Scholar] [CrossRef]
- Majchrzykiewicz, J.A.; Lubelski, J.; Moll, G.N.; Kuipers, A.; Bijlsma, J.J.E.; Kuipers, O.P.; Rink, R. Production of a Class II Two-Component Lantibiotic of Streptococcus Pneumoniae Using the Class I Nisin Synthetic Machinery and Leader Sequence. Antimicrob. Agents Chemother. 2010, 54, 1498–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kautsar, S.A.; van der Hooft, J.J.J.; de Ridder, D.; Medema, M.H. BiG-SLiCE: A Highly Scalable Tool Maps the Diversity of 1.2 Million Biosynthetic Gene Clusters. Gigascience 2021, 10, giaa154. [Google Scholar] [CrossRef] [PubMed]
- Kautsar, S.A.; Blin, K.; Shaw, S.; Weber, T.; Medema, M.H. BiG-FAM: The Biosynthetic Gene Cluster Families Database. Nucleic Acids Res. 2021, 49, D490–D497. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. AntiSMASH 5.0: Updates to the Secondary Metabolite Genome Mining Pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]
- Mungan, M.D.; Alanjary, M.; Blin, K.; Weber, T.; Medema, M.H.; Ziemert, N. ARTS 2.0: Feature Updates and Expansion of the Antibiotic Resistant Target Seeker for Comparative Genome Mining. Nucleic Acids Res. 2020, 48, W546–W552. [Google Scholar] [CrossRef]
- Mungan, M.D.; Blin, K.; Ziemert, N. ARTS-DB: A Database for Antibiotic Resistant Targets. Nucleic Acids Res. 2022, 50, D736–D740. [Google Scholar] [CrossRef]
- De los Santos, E.L.C. NeuRiPP: Neural Network Identification of RiPP Precursor Peptides. Sci. Rep. 2019, 9, 13406. [Google Scholar] [CrossRef] [Green Version]
- Hannigan, G.D.; Prihoda, D.; Palicka, A.; Soukup, J.; Klempir, O.; Rampula, L.; Durcak, J.; Wurst, M.; Kotowski, J.; Chang, D.; et al. A Deep Learning Genome-Mining Strategy for Biosynthetic Gene Cluster Prediction. Nucleic Acids Res. 2019, 47, e110. [Google Scholar] [CrossRef]
- Mohimani, H.; Kersten, R.D.; Liu, W.-T.; Wang, M.; Purvine, S.O.; Wu, S.; Brewer, H.M.; Pasa-Tolic, L.; Bandeira, N.; Moore, B.S.; et al. Automated Genome Mining of Ribosomal Peptide Natural Products. ACS Chem. Biol. 2014, 9, 1545–1551. [Google Scholar] [CrossRef]
- Medema, M.H.; Paalvast, Y.; Nguyen, D.D.; Melnik, A.; Dorrestein, P.C.; Takano, E.; Breitling, R. Pep2Path: Automated Mass Spectrometry-Guided Genome Mining of Peptidic Natural Products. PLoS Comput. Biol. 2014, 10, e1003822. [Google Scholar] [CrossRef]
- Kirkpatrick, C.L.; Broberg, C.A.; McCool, E.N.; Lee, W.J.; Chao, A.; McConnell, E.W.; Pritchard, D.A.; Hebert, M.; Fleeman, R.; Adams, J.; et al. The “PepSAVI-MS” Pipeline for Natural Product Bioactive Peptide Discovery. Anal. Chem. 2017, 89, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Mohimani, H.; Liu, W.-T.; Mylne, J.S.; Poth, A.G.; Colgrave, M.L.; Tran, D.; Selsted, M.E.; Dorrestein, P.C.; Pevzner, P.A. Cycloquest: Identification of Cyclopeptides via Database Search of Their Mass Spectra against Genome Databases. J. Proteome Res. 2011, 10, 4505–4512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila-Farres, X.; Chu, J.; Inoyama, D.; Ternei, M.A.; Lemetre, C.; Cohen, L.J.; Cho, W.; Reddy, B.V.B.; Zebroski, H.A.; Freundlich, J.S.; et al. Antimicrobials Inspired by Nonribosomal Peptide Synthetase Gene Clusters. J. Am. Chem. Soc. 2017, 139, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Vila-Farres, X.; Chu, J.; Ternei, M.A.; Lemetre, C.; Park, S.; Perlin, D.S.; Brady, S.F. An Optimized Synthetic-Bioinformatic Natural Product Antibiotic Sterilizes Multidrug-Resistant Acinetobacter Baumannii-Infected Wounds. mSphere 2018, 3, e00528-17. [Google Scholar] [CrossRef] [Green Version]
- Hudson, G.A.; Zhang, Z.; Tietz, J.I.; Mitchell, D.A.; vander Donk, W.A. In Vitro Biosynthesis of the Core Scaffold of the Thiopeptide Thiomuracin. J. Am. Chem. Soc. 2015, 137, 16012–16015. [Google Scholar] [CrossRef]
- DiCaprio, A.J.; Firouzbakht, A.; Hudson, G.A.; Mitchell, D.A. Enzymatic Reconstitution and Biosynthetic Investigation of the Lasso Peptide Fusilassin. J. Am. Chem. Soc. 2019, 141, 290–297. [Google Scholar] [CrossRef]
- Mahanta, N.; Liu, A.; Dong, S.; Nair, S.K.; Mitchell, D.A. Enzymatic Reconstitution of Ribosomal Peptide Backbone Thioamidation. Proc. Natl. Acad. Sci. USA 2018, 115, 3030. [Google Scholar] [CrossRef] [Green Version]
- Koos, J.D.; Link, A.J. Heterologous and in Vitro Reconstitution of Fuscanodin, a Lasso Peptide from Thermobifida Fusca. J. Am. Chem. Soc. 2019, 141, 928–935. [Google Scholar] [CrossRef]
- Chevrette, M.G.; Carlson, C.M.; Ortega, H.E.; Thomas, C.; Ananiev, G.E.; Barns, K.J.; Book, A.J.; Cagnazzo, J.; Carlos, C.; Flanigan, W.; et al. The Antimicrobial Potential of Streptomyces from Insect Microbiomes. Nat. Commun. 2019, 10, 516. [Google Scholar] [CrossRef] [Green Version]






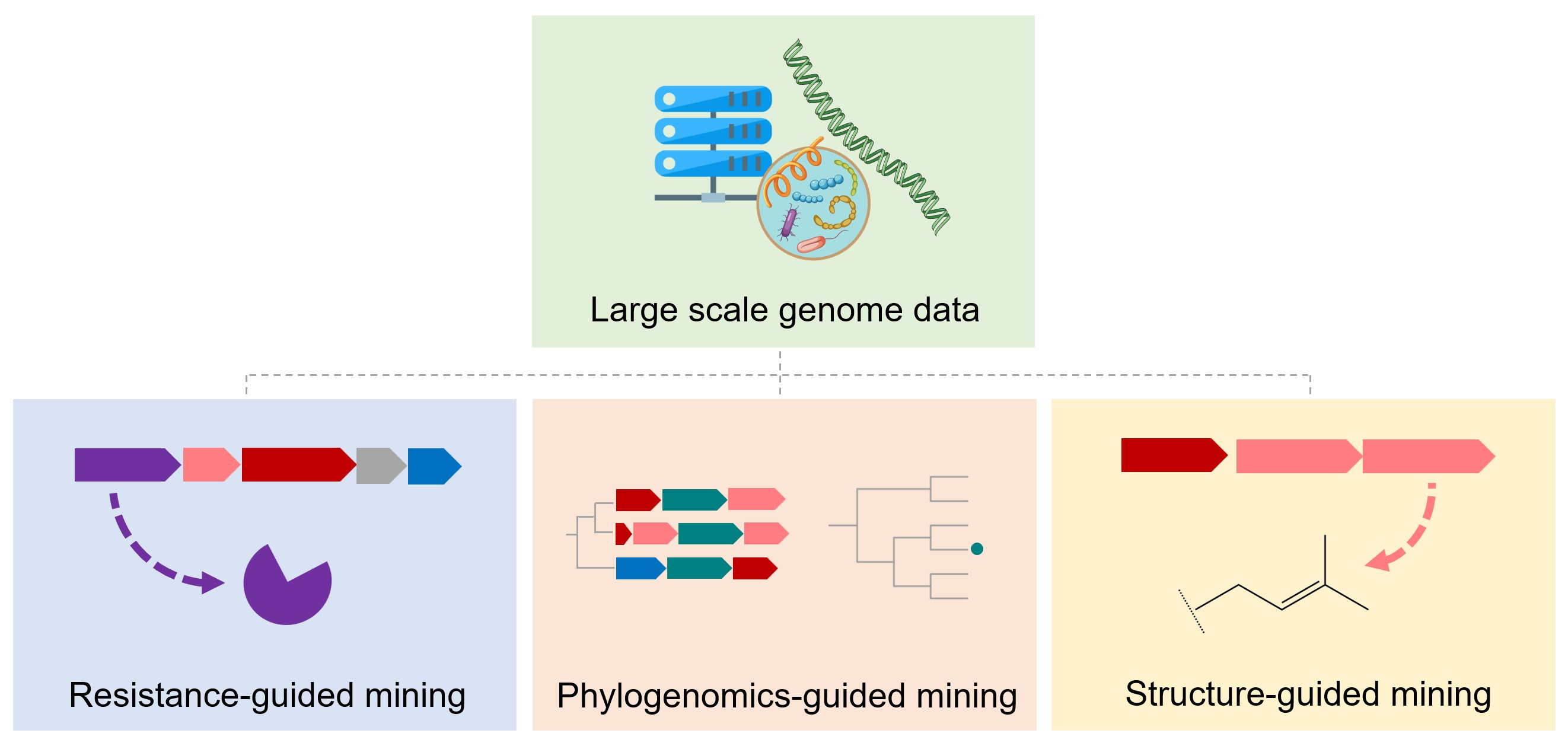{kind=link}
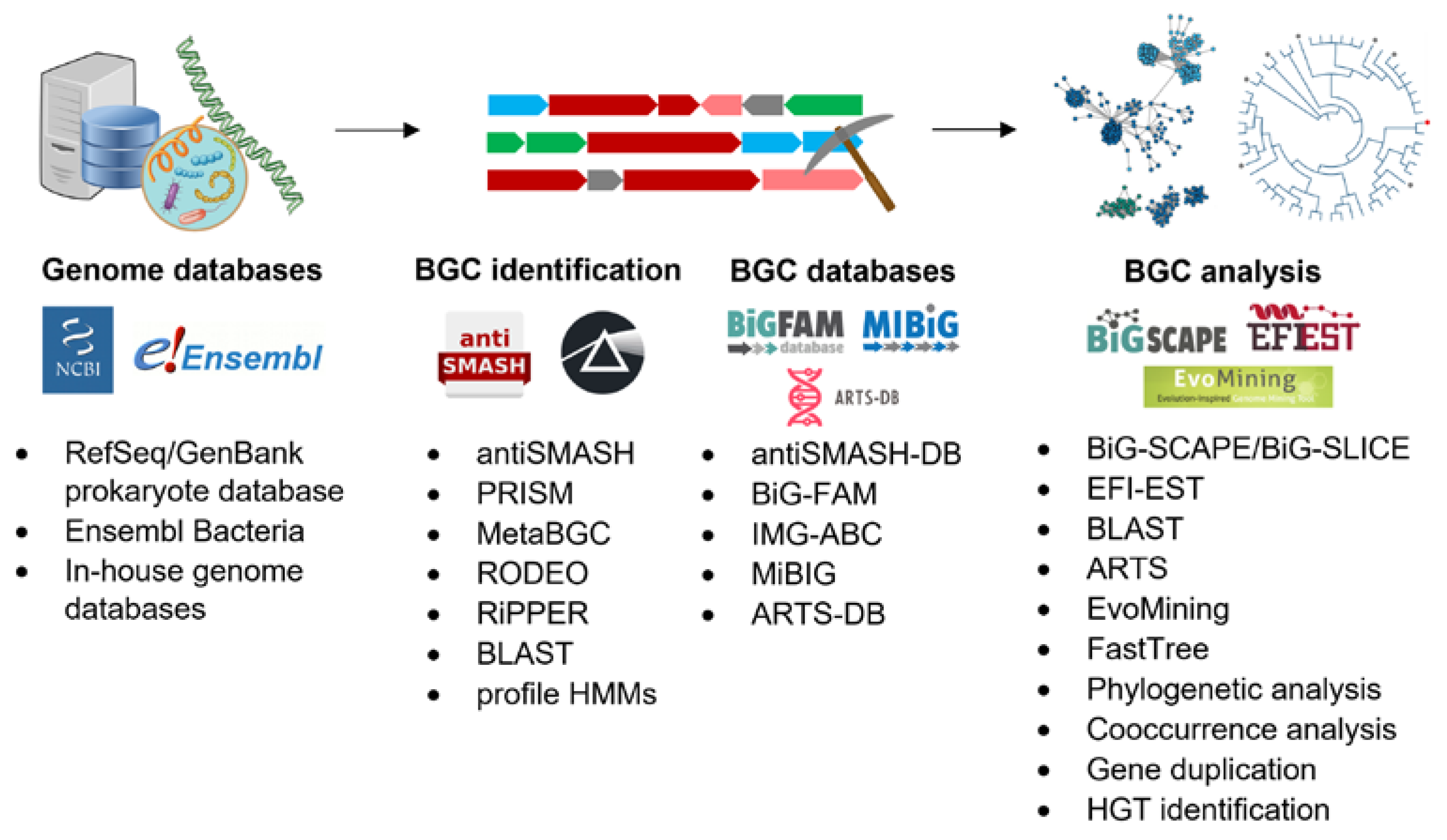{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Resistance-Gene-Guided | |||
|---|---|---|---|
| Resistance Gene(s) | Natural Product | Source Organism | Reference |
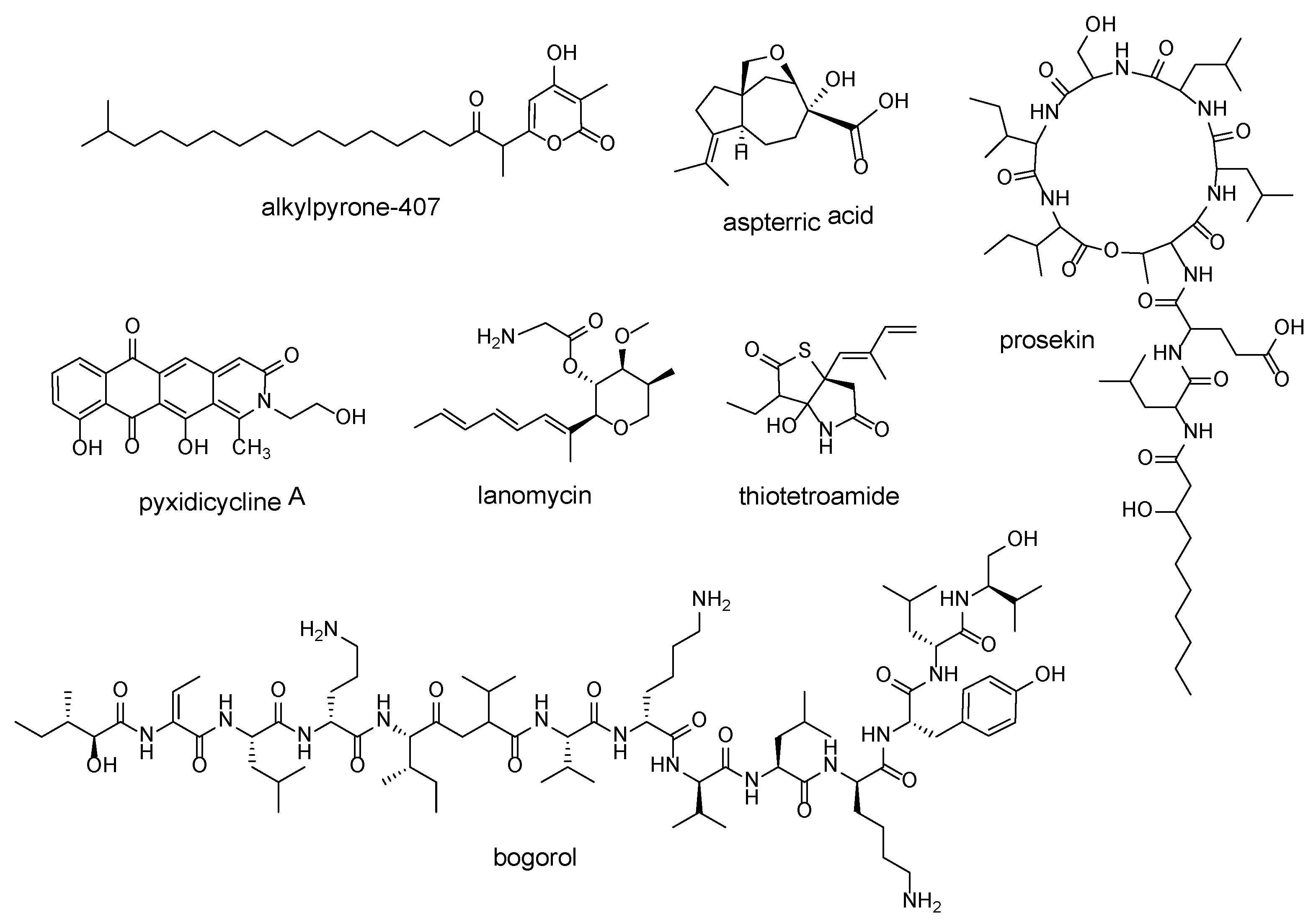
| pentapeptide repeat protein (PRP) sequences | alkylpyrone-407 | Cystobacterineae strain MCy9487 | [39] |
| pyxidicycline A | Pyxidicoccus fallax An d48 | [40] | |
| dihydroxyacid dehydratase | aspterric acid | Aspergillus terreus NIH2624 | [41] |
| tripartite efflux system PleABC | prosekin | Pseudomonas prosekii LMG 26867 | [42] |
| lanosterol 14α-demethylase | lanomycin | Pyrenophora dematioidea TTI-1096 | [43] |
| fatty acid synthase | thiotetroamide | Streptomyces afghaniensis NRRL 5621 | [44] |
| D-stereospecific peptidase | bogorol | Brevibacillus laterosporus DSM 25 | [45] |
| Phylogenomics-Guided | |||
| Sequences Used for Phylogenetic Analysis | Natural Product | Source Organism | Reference |
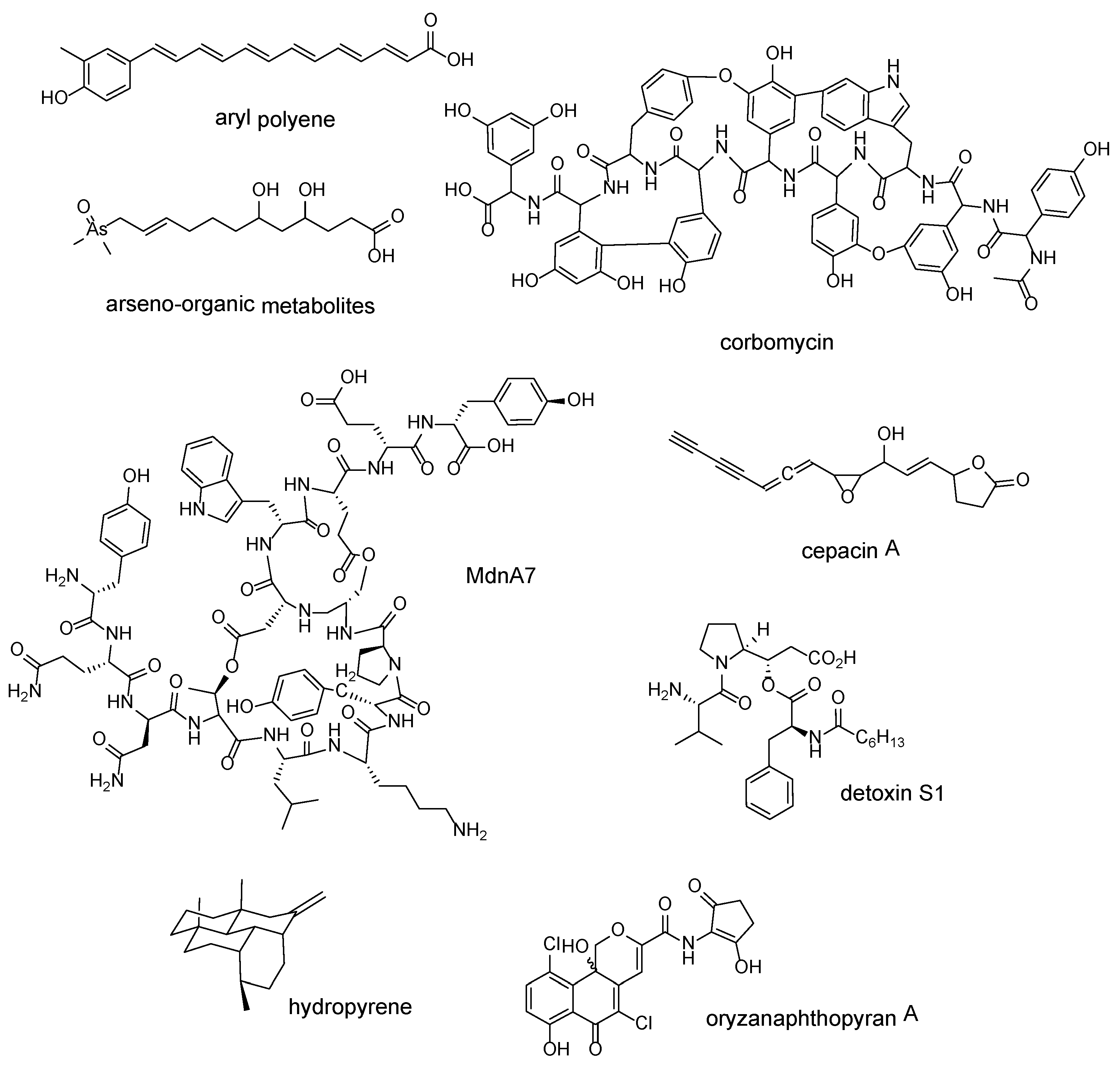
| Whole BGCs of different families | aryl polyenes | Escherichia coli CFT073 | [46] |
| “Expanded-then-recruited” enzyme families; 3-carboxyvinyl-phosphoshikimate transferase | arseno-organic metabolites | Streptomyces lividans 66 | [47] |
| Each shared gene found in glycopeptide antibiotic-producing BGCs | corbomycin | Streptomyces sp. WAC01529 | [48] |
| ATP-grasp ligase | MdnA7 | Cyanothece sp. PCC 7822 | [49] |
| LuxR | cepacin A | Burkholderia ambifaria BCC0191 | [50] |
| Whole BGCs containing tauD expansion | detoxin S1 | Streptomyces sp. NRRL S-325 | [51] |
| terpene synthase | hydropyrene | Streptomyces clavuligerus ATCC 27064 | [52] |
| chain length factor (CLF) protein | oryzanaphthopyran A | Streptacidiphilus oryzae CGMCC 4.2012 | [53] |
| Structure-Guided | |||
| Targeted Chemical Structure | Natural Product | Source Organism | Reference |
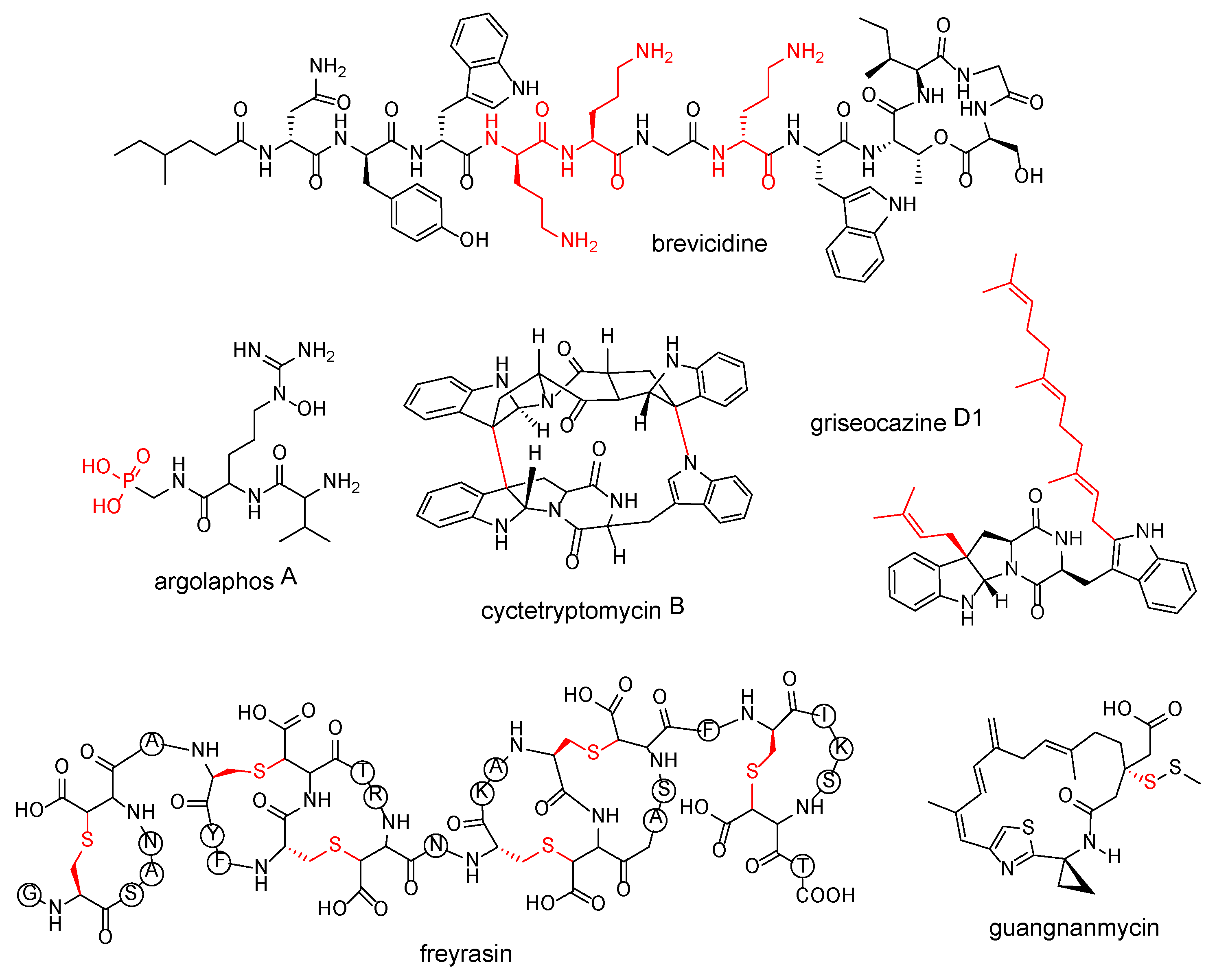
| cationic amino acid residues | brevicidine | Brevibacillus laterosporus DSM 25 | [54] |
| chemical transformations catalyzed by cytochrome P450 on cyclodipeptides | cyctetryptomycin B | Saccharopolyspora hirsuta DSM 44795 | [55] |
| prenyl groups on cyclodipeptides | griseocazine D1 | Streptomyces griseocarneus 132 | [56] |
| thioether bonds | freyrasin | Paenibacillus polymyxa ATCC 842 | [57] |
| chemical transformations catalyzed by the DUF–SH didomain | guangnanmycin | Streptomyces sp. CB01883 | [58] |
| phosphonic acid | argolaphos A | Streptomyces monomycini NRRLB-24309 | [59] |
| Global Genome Mining for RiPPs | |||
| Combining the structure-guided strategy with precursor peptide sequence search | |||
| RiPP Family | Natural Product | Source Organism | Reference |
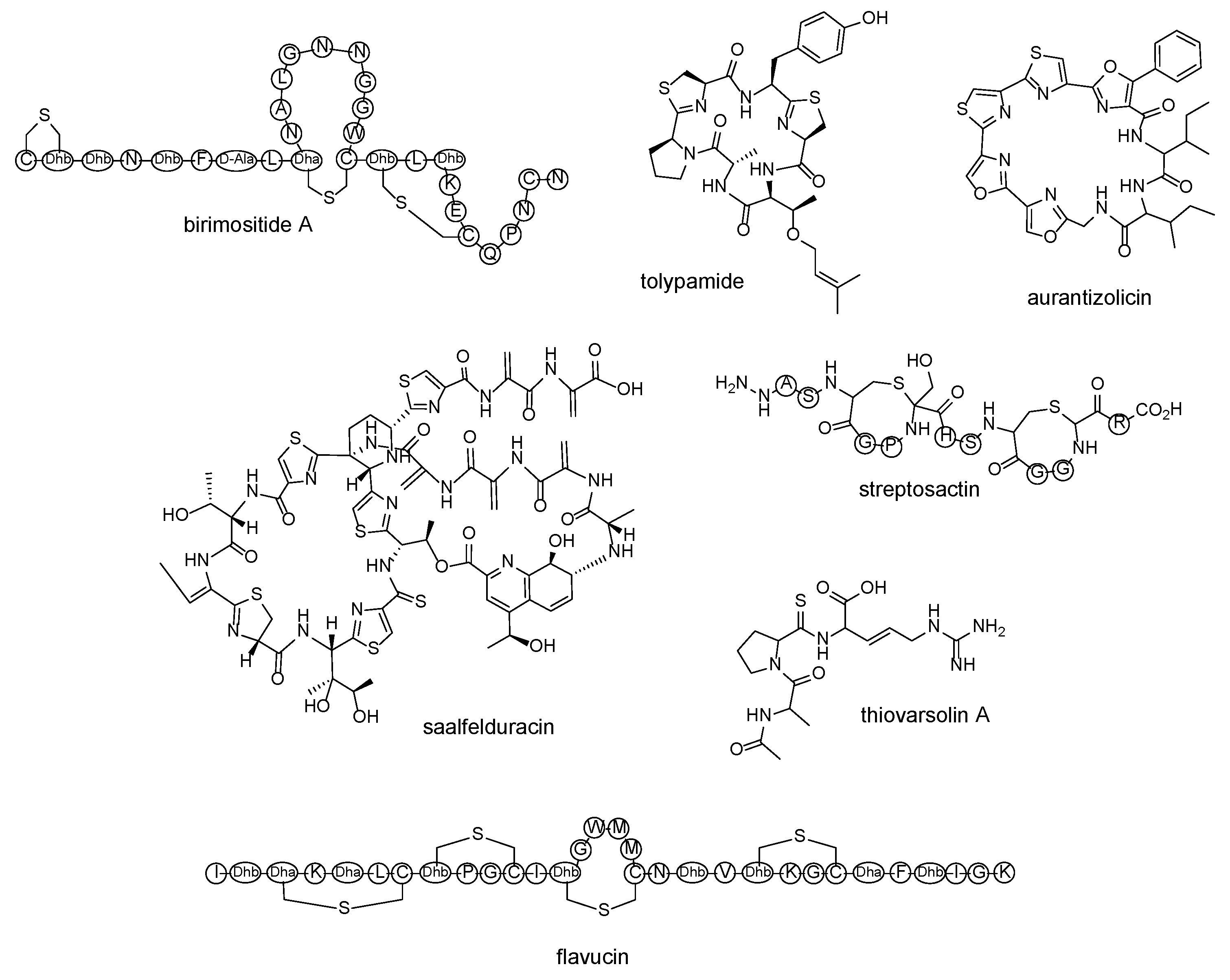
| lanthipeptide | birimositide | Streptomyces rimosus subsp. rimosus WC3908 | [60] |
| cyanobactin | tolypamide | Tolypothrix sp. PCC 7601 | [61] |
| polyoxazole-thiazole-based cyclopeptide | aurantizolicin | Streptomyces auranticaus JA 4570 | [62] |
| thiopeptide | saalfelduracin | Amycolatopsissaalfeldensis NRRL B-24474 | [63] |
| thioamitides | thiovarsolin A | Streptomyces varsoviensis DSM 40346 | [64] |
| sactipeptide | streptosactin | Streptococcus thermophilus JIM 8232 | [65] |
| lanthipeptide | flavucin, agalacticin, etc. | Corynebacterium lipophiloflavum DSM 44291 | [66] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malit, J.J.L.; Leung, H.Y.C.; Qian, P.-Y. Targeted Large-Scale Genome Mining and Candidate Prioritization for Natural Product Discovery. Mar. Drugs 2022, 20, 398. https://doi.org/10.3390/md20060398
Malit JJL, Leung HYC, Qian P-Y. Targeted Large-Scale Genome Mining and Candidate Prioritization for Natural Product Discovery. Marine Drugs. 2022; 20(6):398. https://doi.org/10.3390/md20060398
Chicago/Turabian StyleMalit, Jessie James Limlingan, Hiu Yu Cherie Leung, and Pei-Yuan Qian. 2022. "Targeted Large-Scale Genome Mining and Candidate Prioritization for Natural Product Discovery" Marine Drugs 20, no. 6: 398. https://doi.org/10.3390/md20060398