
New 3-Acyl Tetramic Acid Derivatives from the Deep-Sea-Derived Fungus Lecanicillium fusisporum

 ,
,
Abstract
:
1. Introduction
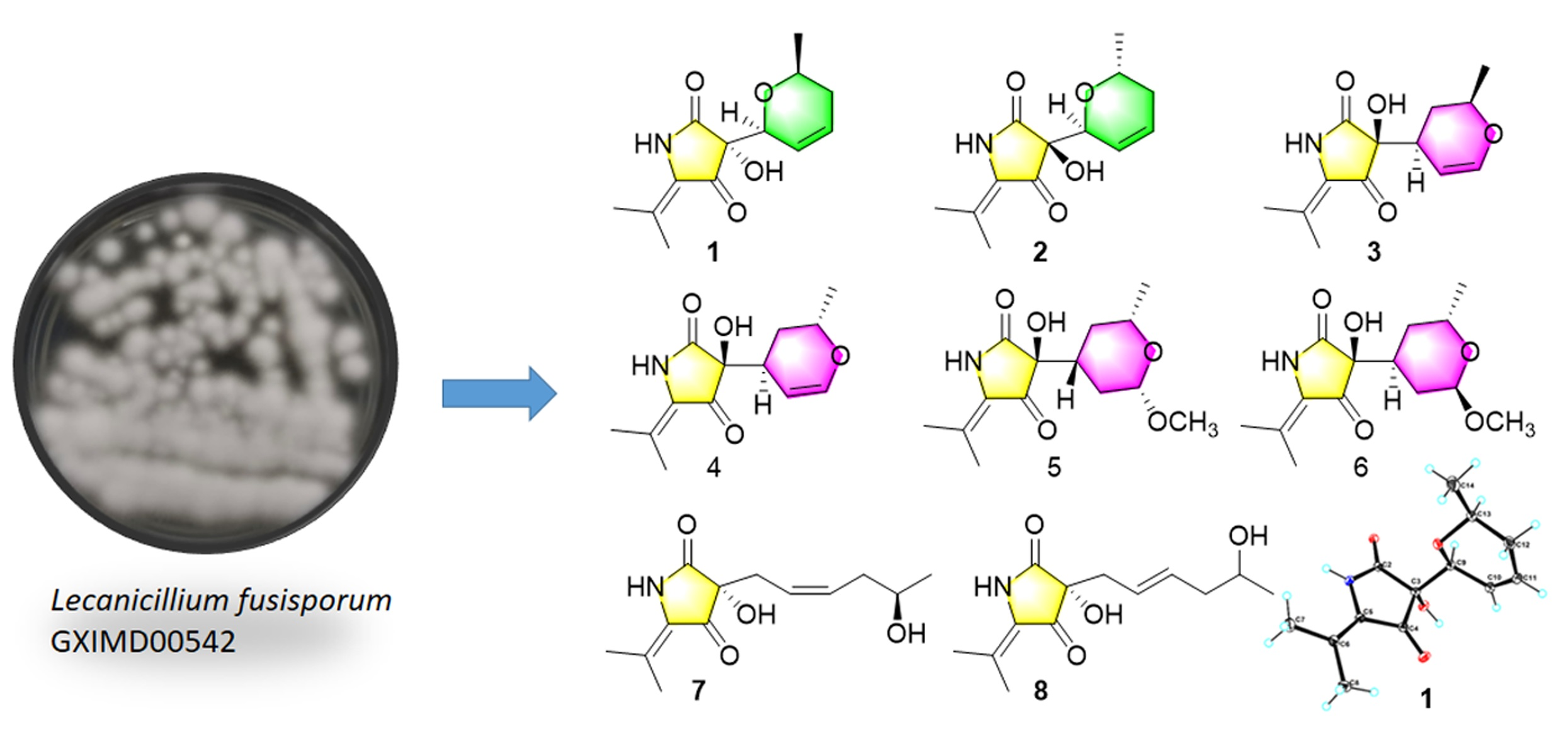
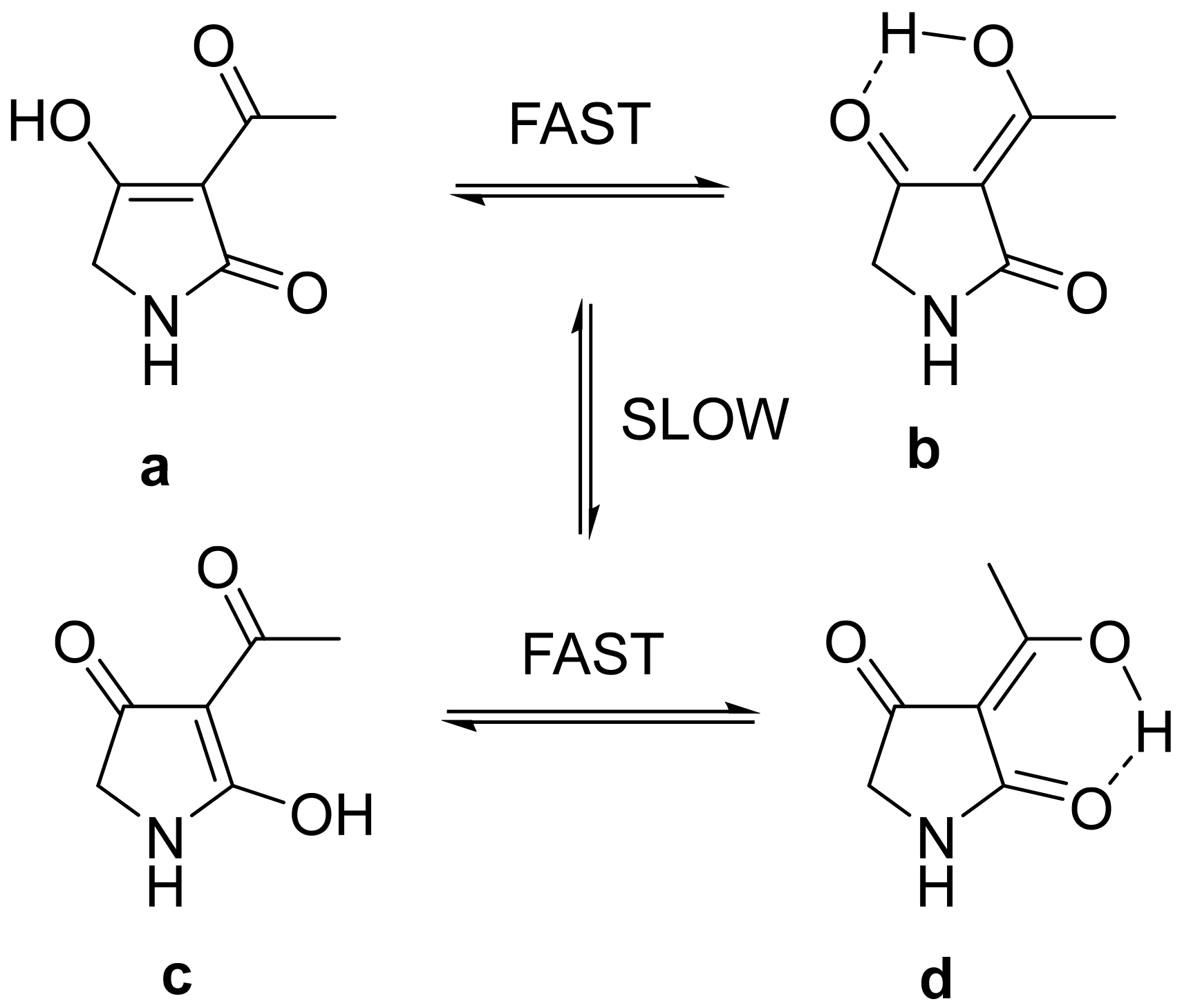
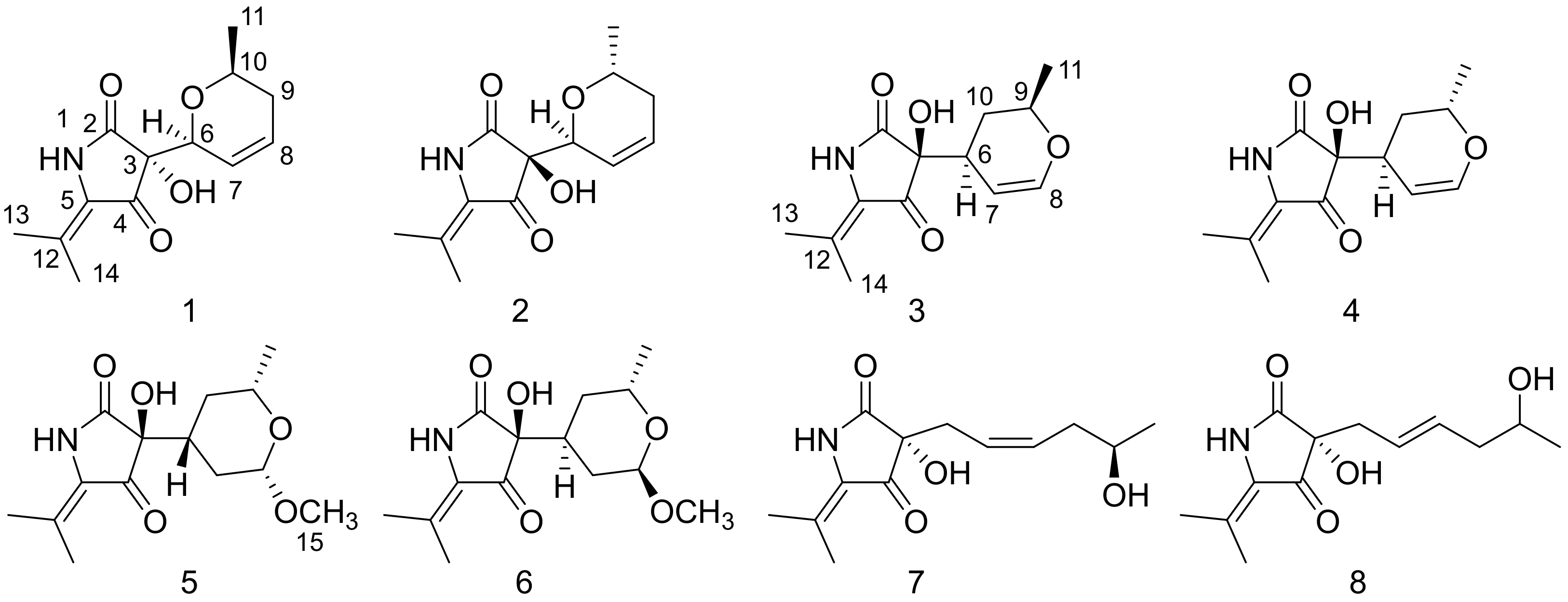
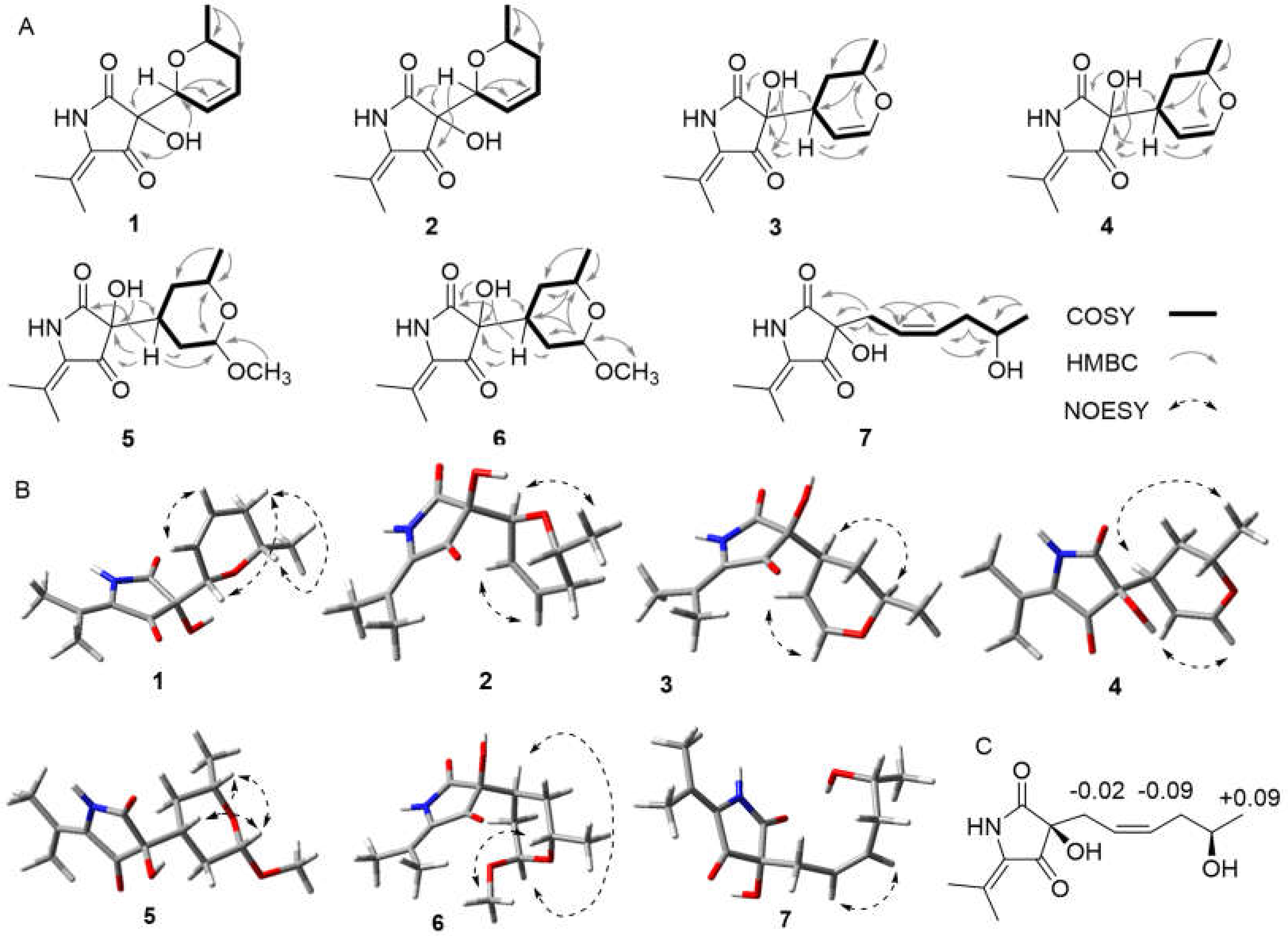
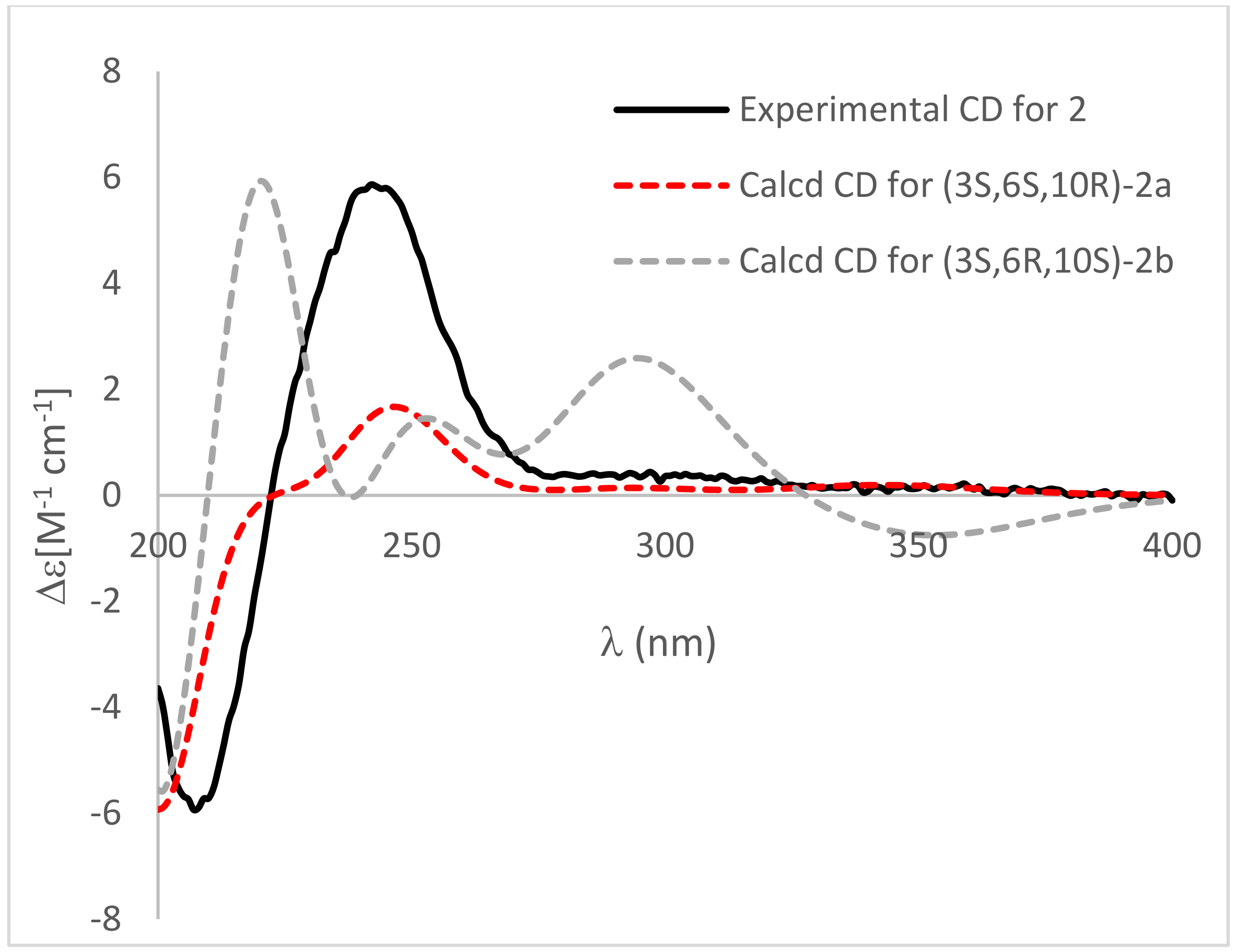
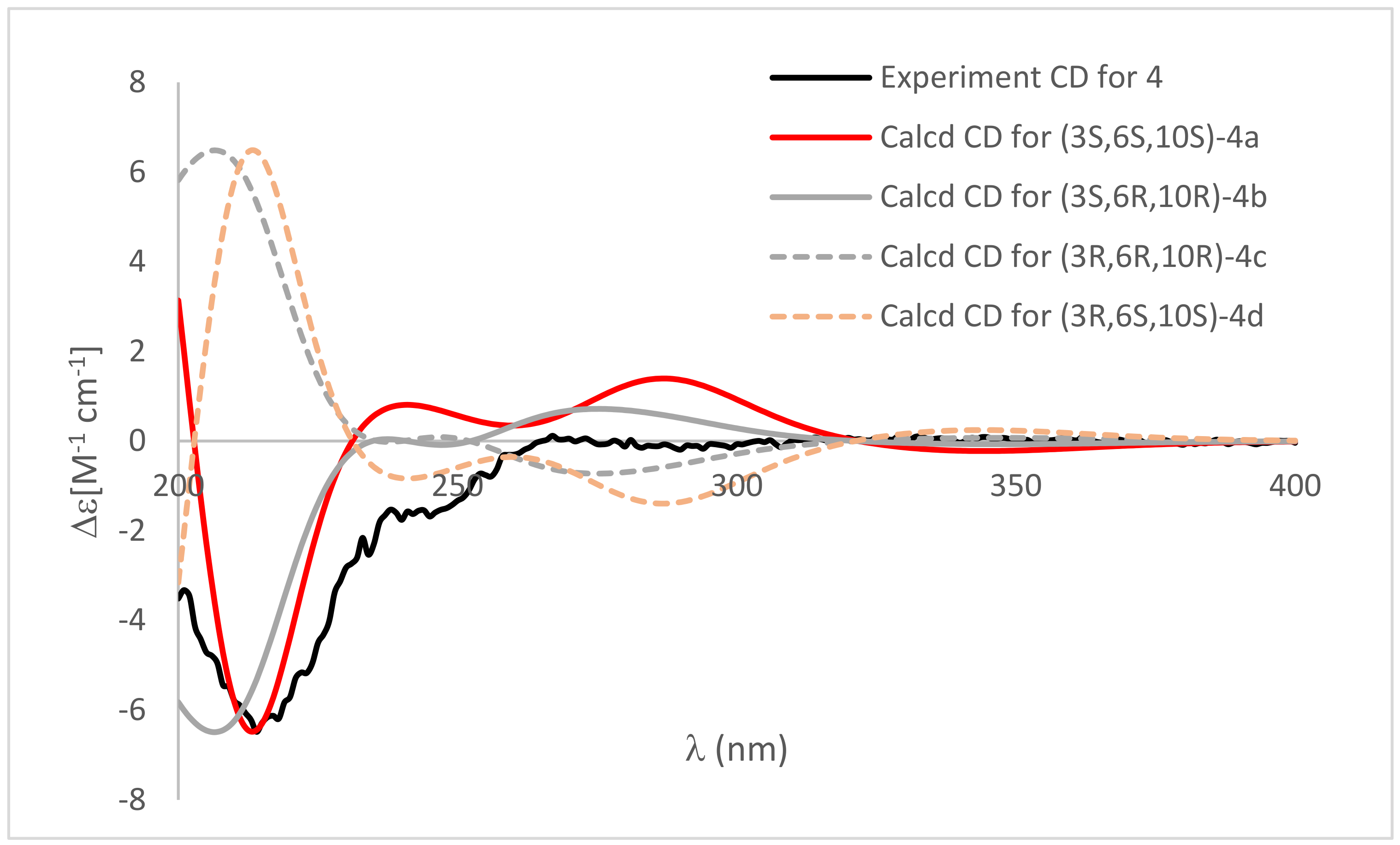
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedure
3.2. Fungal Material
3.3. Fermentation and Extraction
3.4. Isolation and Purification
3.5. Mosher’s Ester Reaction
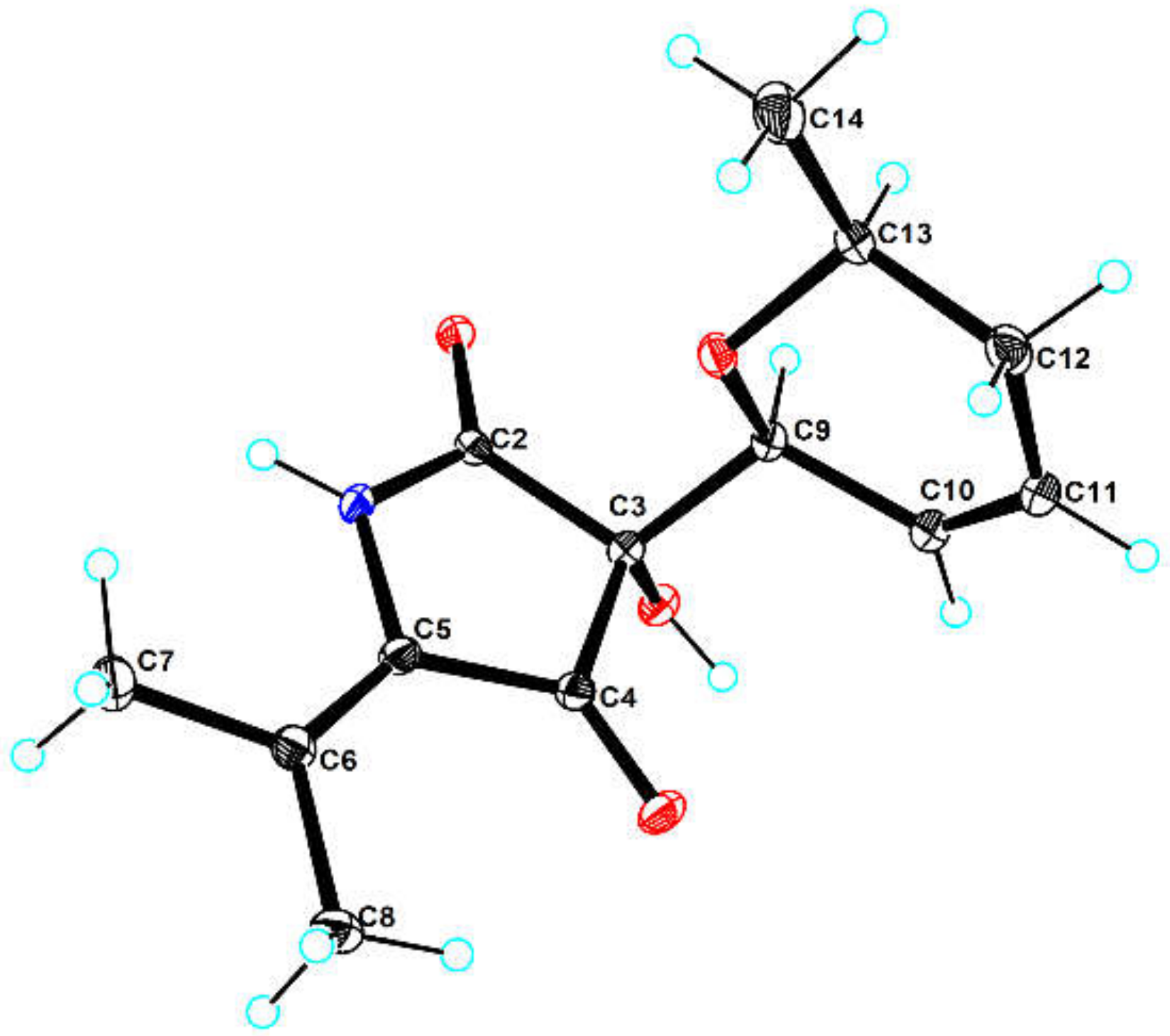
3.6. X-ray Crystal Structure Analysis
3.7. Computational Methods
3.8. Cytotoxicity Assay
3.9. NF-κB Luciferase Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pang, X.; Chen, W.; Wang, X.; Zhou, X.; Yang, B.; Tian, X.; Wang, J.; Xu, S.; Liu, Y. New tetramic acid derivatives from the deep-sea-derived fungus Penicillium sp. SCSIO06868 with SARS-CoV-2 M(pro) inhibitory activity evaluation. Front. Microbiol. 2021, 12, 730807. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.; Gulder, T. Biosynthetic strategies for tetramic acid formation. Nat. Prod. Rep. 2021, 38, 1555–1566. [Google Scholar] [CrossRef]
- Ghisalberti, E.L. Bioactive Tetramic Acid Metabolites. In Studies in Natural Products Chemistry; Rahman, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 28, pp. 109–163. [Google Scholar] [CrossRef]
- Jiang, M.; Chen, S.; Li, J.; Liu, L. The biological and chemical diversity of tetramic acid compounds from marine-derived microorganisms. Mar. Drugs 2020, 18, 114. [Google Scholar] [CrossRef] [Green Version]
- Royles, B.J.L. Naturally Occurring Tetramic Acids: Structure, Isolation, and Synthesis. Chem. Rev. 1995, 95, 1981–2001. [Google Scholar] [CrossRef]
- Schobert, R.; Schlenk, A. Tetramic and tetronic acids: An update on new derivatives and biological aspects. Bioorg. Med. Chem. 2008, 16, 4203–4221. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.; Li, Q.; Ju, J. Naturally occurring tetramic acid products: Isolation, structure elucidation and biological activity. RSC Adv. 2014, 4, 50566–50593. [Google Scholar] [CrossRef]
- Huang, Z.; Nong, X.; Liang, X.; Qi, S. New tetramic acid derivatives from the deep-sea-derived fungus Cladosporium sp. SCSIO z0025. Tetrahedron 2018, 74, 2620–2626. [Google Scholar] [CrossRef]
- Zare, R.; Gams, W. A revision of Verticillium section Prostrata. IV. The genera Lecanicillium and Simplicillium gen. nov. Nova Hedwigia 2001, 73, 1–50. [Google Scholar] [CrossRef]
- Steenberg, T.; Humber, R.A. Entomopathogenic potential of Verticillium and Acremonium species (Deuteromycotina: Hyphomycetes). J. Invertebr. Pathol. 1999, 73, 309–314. [Google Scholar] [CrossRef]
- Ekbom, B.S.; Ahman, I. The fungus Verticillium fusisporum as an insect pathogen. J. Invertebr. Pathol. 1980, 36, 136–138. [Google Scholar] [CrossRef]
- Miller, L.K.; Lingg, A.J.; Bulla, L.A., Jr. Bacterial, viral, and fungal insecticides. Science 1983, 219, 715–721. [Google Scholar] [CrossRef]
- Ishidoh, K.; Kinoshita, H.; Igarashi, Y.; Ihara, F.; Nihira, T. Cyclic lipodepsipeptides verlamelin A and B, isolated from entomopathogenic fungus Lecanicillium sp. J. Antibiot. 2014, 67, 459–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Li, H.J.; Huang, C.B.; Wang, Z.H.; Lan, W.J.; Wang, L.Y. Alkaloids from the marine fungus Lecanicillium fusisporum using an amino acid-directed strategy. Nat. Prod. Commun. 2021, 16, 1–6. [Google Scholar] [CrossRef]
- Zanardi, M.M.; Sarotti, A.M. Sensitivity analysis of DP4+ with the probability distribution terms: Development of a universal and customizable method. J. Org. Chem. 2021, 86, 8544–8548. [Google Scholar] [CrossRef]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. 2015, 80, 12–34. [Google Scholar] [CrossRef]
- Pansanit, A.; Park, E.J.; Kondratyuk, T.P.; Pezzuto, J.M.; Lirdprapamongkol, K.; Kittakoop, P. Vermelhotin, an anti-inflammatory agent, suppresses nitric oxide production in RAW 264.7 cells via p38 inhibition. J. Nat. Prod. 2013, 76, 1824–1827. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Wang, S.; Wang, G.; Pang, K.; Lee, C.; Kuo, Y.; Cha, H.; Lin, R.; Lee, T. Angiogenesis inhibitors and anti-inflammatory agents from Phoma sp. NTOU4195. J. Nat. Prod. 2016, 79, 2983–2990. [Google Scholar] [CrossRef]
- Xu, X.; Tsang, S.W.; Guan, Y.; Liu, K.; Pan, W.; Lam, C.S.; Lee, K.M.; Xia, Y.; Xie, W.; Wong, W.Y.; et al. In vitro and in vivo antitumor effects of plant-derived miliusanes and their induction of cellular senescence. J. Med. Chem. 2019, 62, 1541–1561. [Google Scholar] [CrossRef]
- Spartan’14; Wavefunction Inc.: Irvine, CA, USA, 2013.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Rev. B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bruhn, T.; Schaumloffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Deng, W.; Zhang, Y.; Ke, M.; Zou, B.; Luo, X.; Su, J.; Wang, Y.; Xu, J.; Nandakumar, K.S.; et al. A marine fungus-derived nitrobenzoyl sesquiterpenoid suppresses receptor activator of NF-kappaB ligand-induced osteoclastogenesis and inflammatory bone destruction. Br. J. Pharmacol. 2020, 177, 4242–4260. [Google Scholar] [CrossRef]
- Rischer, M.; Lee, S.R.; Eom, H.J.; Park, H.B.; Vollmer, J.; Kaster, A.K.; Shin, Y.H.; Oh, D.C.; Kim, K.H.; Beemelmanns, C. Spirocyclic cladosporicin A and cladosporiumins I and J from a Hydractinia-associated Cladosporium sphaerospermum SW67. Org. Chem. Front. 2019, 6, 1084–1093. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | 7 |
|---|---|---|---|
| 6 | 4.30, br s | 4.23, m | 2.41, dd, (14.1, 7.5) |
| 7 | 5.97, d, (10.4) | 5.95, m | 5.17, dt, (10.9, 7.3) |
| 8 | 5.82, dtd, (10.2, 2.1, 5.7) | 5.79, dtd, (9.9, 4.3, 1.9) | 5.49, dt, (10.9, 7.3) |
| 9 | 1.90, m, H-9α | 1.93, m, | 1.94–2.04, ovp. |
| 1.65, m, H-9β | 1.74, ovp. a, | ||
| 10 | 3.52, ddd, (15.6, 6.1, 4.6) | 3.52, dtt, (12.3, 6.1, 3.5) | 3.52, m |
| 11 | 0.99, d, (6.1) | 0.99, d, (6.1) | 0.98, d, (6.2) |
| 13 | 1.77, s | 1.77, s | 1.78, s |
| 14 | 2.02, s | 2.07, s | 2.06, s |
| NH-1 | 10.33, br s | 10.19, br s | 10.34, br s |
| OH-3 | 6.32, br s | 6.35, br s | 6.14, br s |
| OH-13 | - | - | 4.44, d, (4.5) |
| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| 2 | 171.45, C | 169.7, C | 171.35, C | 171.89, C | 171.51, C | 171.53, C | 171.91, C |
| 3 | 74.31, C | 74.22, C | 75.14, C | 75.85, C | 75.29, C | 75.55, C | 73.96, C |
| 4 | 197.12, C | 199.36, C | 199.04, C | 199.64, C | 199.59, C | 199.65, C | 199.38, C |
| 5 | 120.24, C | 120.52, C | 122.3, C | 122.07, C | 122.44, C | 122.28, C | 122.47, C |
| 6 | 76.97, CH | 77.68, CH | 37.73, CH | 34.26, CH | 39.6, CH | 35.23, CH | 33.46, CH2 |
| 7 | 125.37, CH | 125.44, CH | 97.81, CH | 96.65, CH | 30.31, CH2 | 28.86, CH2 | 121.66, CH |
| 8 | 124.85, CH | 124.85, CH | 145.33, CH | 145.25, CH | 101.66, CH | 97.05, CH | 130.96, CH |
| 9 | 32.02, CH2 | 32.02, CH2 | 71.2, CH | 68.28, CH | 69.93, CH | 63.82, CH | 36.74, CH2 |
| 10 | 69.62, CH | 69.73, CH | 29.7, CH2 | 27.95, CH2 | 31.72, CH2 | 31.85, CH2 | 65.81, CH |
| 11 | 21.4, CH3 | 21.35, CH3 | 21.17, CH3 | 20.63, CH3 | 21.45, CH3 | 21.6, CH3 | 23.11, CH3 |
| 12 | 128.96, C | 129.13, C | 128.57, C | 128.6, C | 128.5, C | 128.44, C | 128.41, C |
| 13 | 20.57, CH3 | 20.57, CH3 | 20.71, CH3 | 19.91, CH3 | 20.69, CH3 | 20.66, CH3 | 20.69, CH3 |
| 14 | 18.39, CH3 | 18.39, CH3 | 18.49, CH3 | 18.44, CH3 | 18.46, CH3 | 18.43, CH3 | 18.46, CH3 |
| 15 | - | - | - | - | 55.32, CH3 | 53.67, CH3 | - |
| No. | 3 | 4 | 5 | 6 |
|---|---|---|---|---|
| 6 | 2.64, ddt, (11.4, 5.8, 2.0) | 2.45, ddd, (10.5, 4.7, 2.4) | 1.98, tt, (12.5, 3.7) | 2.17, tt, (12.8, 3.5) |
| 7 | 4.51, d, (6.4) | 4.44, dd, (6.4, 3.4) | 1.59, dt, (12.5, 2.1) | 1.56, ovp. |
| 1.05, td, (12.5, 9.5) | 1.05, ovp. | |||
| 8 | 6.35, dd, (6.4, 2.3) | 6.38, dd, (6.4, 2.2) | 4.23, dd, (9.5, 2.1) | 4.66, d, 2.6 |
| 9 | 3.85, dqd, (12.6, 6.3, 1.7) | 3.91, ddd, (11.5, 6.3, 3.2) | 3.41, dqd, (12.3, 6.1, 1.9) | 3.63, dqd, (12.2, 6.3, 2.1) |
| 10 | 1.78, ovp. a | 2.12, ddd, 1(4.2, 7.0, 3.5) | 1.52, m | 1.56, ovp. |
| 1.53, dt, (13.2, 11.4) | 1.45, ddd, (14.2, 7.0, 3.5) | 0.97, td, (12.7, 11.0) | 1.05, ovp. | |
| 11 | 1.18, d, (6.3) | 1.13, d, (6.3) | 1.11, d, (6.1) | 1.05, d, (6.3) |
| 13 | 1.81, s | 1.78, s | 1.80 s | 1.79, s |
| 14 | 2.09, s | 2.05, s | 2.08, s | 2.07, s |
| 15 | - | - | 3.30, s | 3.19, s |
| NH-1 | 10.37, br s | 10.41, br s | 10.4, br s | 10.37, br s |
| OH-3 | 6.09, br s | 6.07, br s | 6.04, br s | 5.97, br s |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Tan, Y.; Gao, C.; Liu, K.; Tang, Z.; Lu, C.; Li, H.; Zhang, X.; Liu, Y. New 3-Acyl Tetramic Acid Derivatives from the Deep-Sea-Derived Fungus Lecanicillium fusisporum. Mar. Drugs 2022, 20, 255. https://doi.org/10.3390/md20040255
Xu X, Tan Y, Gao C, Liu K, Tang Z, Lu C, Li H, Zhang X, Liu Y. New 3-Acyl Tetramic Acid Derivatives from the Deep-Sea-Derived Fungus Lecanicillium fusisporum. Marine Drugs. 2022; 20(4):255. https://doi.org/10.3390/md20040255
Chicago/Turabian StyleXu, Xinya, Yanhui Tan, Chenghai Gao, Kai Liu, Zhenzhou Tang, Chunju Lu, Haiyan Li, Xiaoyong Zhang, and Yonghong Liu. 2022. "New 3-Acyl Tetramic Acid Derivatives from the Deep-Sea-Derived Fungus Lecanicillium fusisporum" Marine Drugs 20, no. 4: 255. https://doi.org/10.3390/md20040255