2. Results and Discussion
The acetone extract of the soft coral S. ornata was partitioned between Et2O and H2O. The Et2O-soluble portion was subjected to repeated chromatography over silica gel, Sephadex LH-20, and RP-HPLC to afford four new compounds, compounds 1 (11.6 mg), 2 (1.2 mg), 3 (2.0 mg), 4 (12.3 mg) and 5 (17.0 mg), respectively.
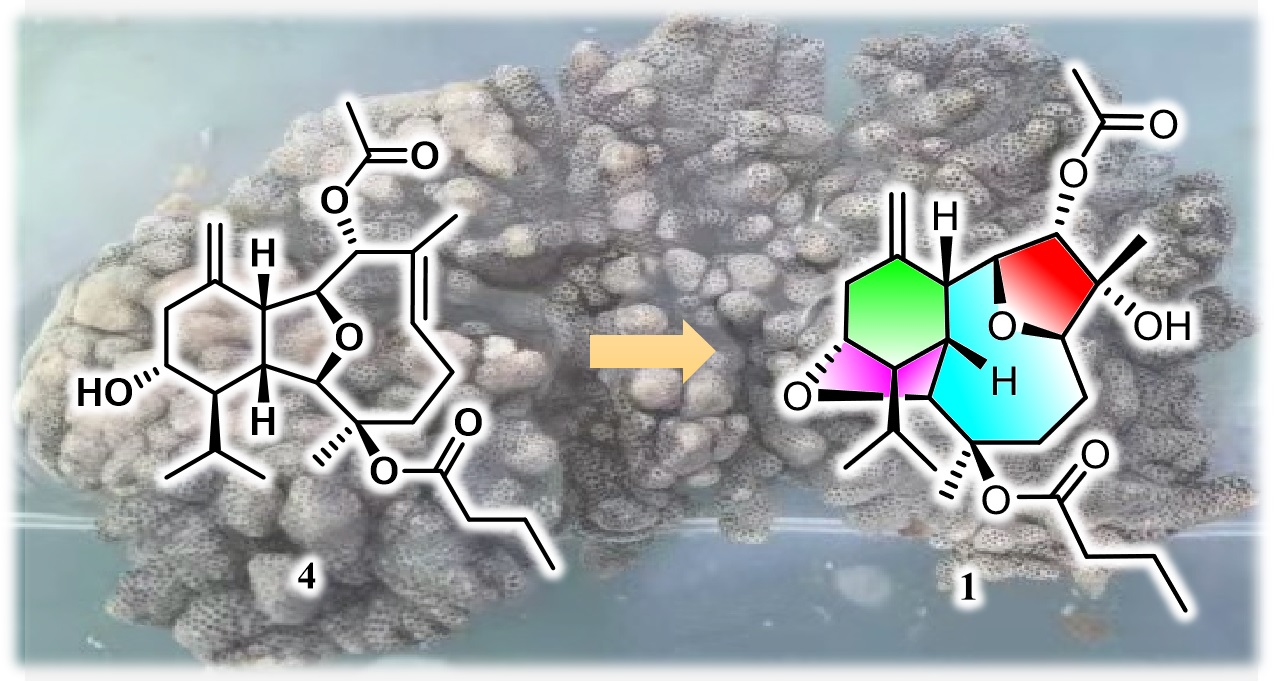
Compound
1 was isolated as an optically active colorless crystal. Mp. 166.2–166.4 °C. Its molecular formula was deduced to be C
26H
40O
7 by the HRESIMS 487.2670 [M + Na]
+ (cald 487.2666), indicating the presence of seven degrees of unsaturation. The
13C NMR, DEPT and HSQC spectra (
Figures S2 and S3) disclosed 26 carbon signals, including six methyls, five sp
3 methylenes, nine sp
3 methines (five oxygenated ones at
δC 75.7, 78.4, 78.9, 86.4, and 86.8), two oxygenated sp
3 quaternary carbons (
δC 79.1, 85.2), one sp
2 methylene, three sp
2 quaternary carbon (two ester carbonyls at
δC 170.2, 173.0). The diagnostic
1H and
13C NMR resonances (
Figures S1 and S2), as well as coupling constants of the connected protons, revealed the presence of one disubstituted terminal double bond (
δH 5.08, s, 1H, H-17a;
δH 5.05, s, 1H, H-17b;
δC 111.5, CH
2, C-17; 144.6, C, C-11). One double bond and two ester carbonyls accounted for three of the total seven degrees of unsaturation, implying a tetracyclic ring system in the molecule.
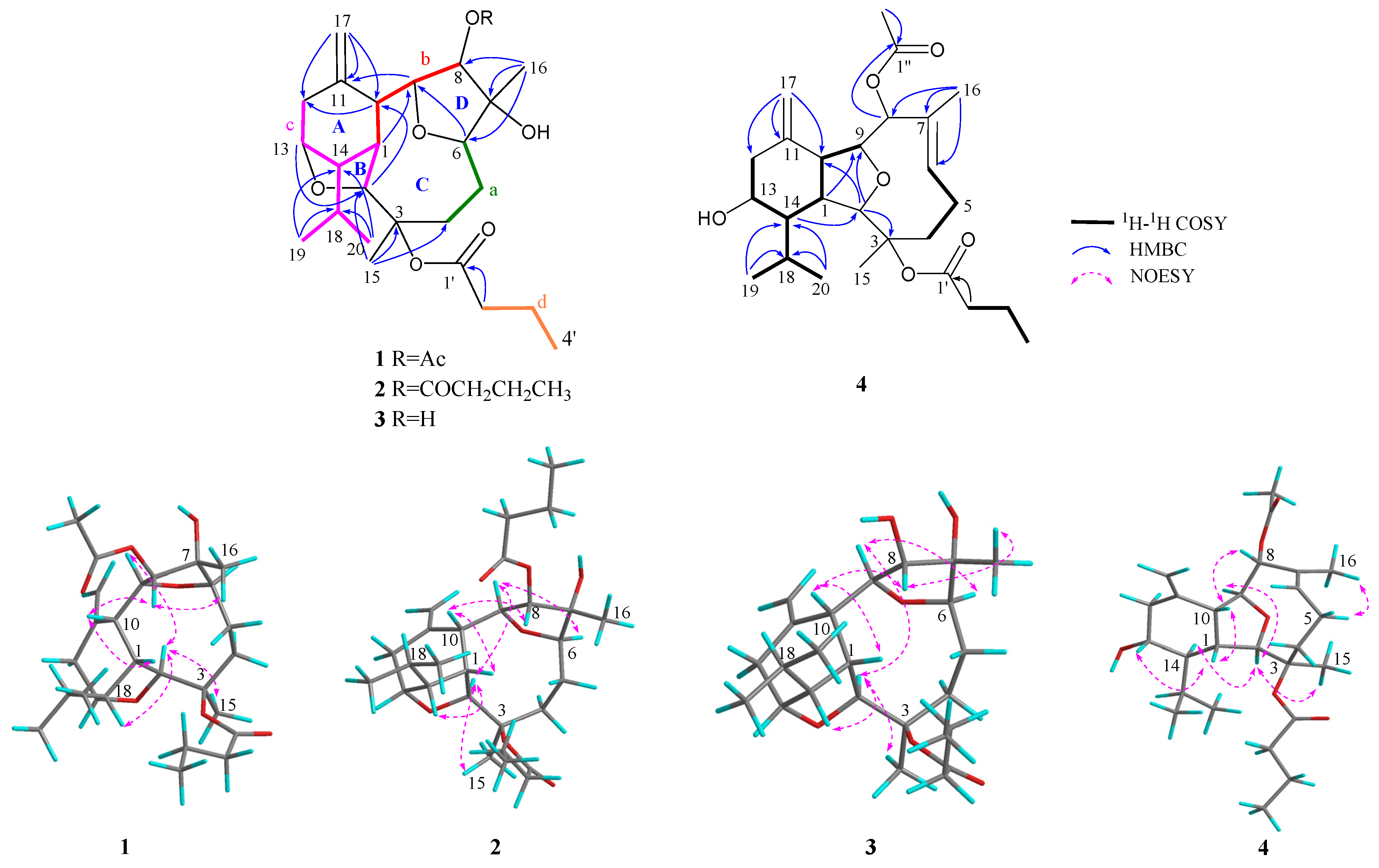
The structure of
1 was established by detailed 2D NMR analysis (
Figure 2). The extensive analysis of the
1H−
1H COSY spectrum (
Figure S5) of
1 elucidated four structural fragments
a–
d, by clear correlations of H
2-4 (
δH 3.01, 1.45)/H
2-5 (
δH 1.50, 1.44)/H-6 (
δH 3.90) (
a); H-8 (
δH 5.27)/H-9 (
δH 4.62)/H-10 (
δH 2.57)/H-1 (
δH 2.65) (
b); H
2-12 (
δH 2.45, 2.38)/H-13 (
δH 4.10)/H-14 (
δH 1.91)/H-1 (
δH 2.65)/H-2 (
δH 4.39), H-14/H-18 (
δH 1.79)/H
3-19 (
δH 1.07) and H-18/H
3-20 (
δH 1.00) (
c); H
2-2′ (
δH 2.26)/H
2-3′ (
δH 1.66)/H
3-4′ (
δH 0.97) (
d), respectively. Fragments
a and
b were deduced to be connected through C-7 by the HMBC correlations (
Figure S4) from H
3-16 to C-6/C7/C-8. The HMBC correlations from H
2-17 to C-10/C-11/C-12 and from H-10 to C-1/C-2/C-12 determined the presence of a cyclohexane ring (ring A) with a terminal double bond at C-11 and an isopropyl group at C-14. The cross peaks from H
3-15 to C-2/C-3/C-4, revealed that the fragments
a and
c were connected via the quaternary carbon C-3. Furthermore, the presence of ether bridges between C-2 and C-13 and between C-6 and C-9 were deduced by HMBC correlations from H-13 to C-2 and from H-6 to C-9, respectively, forming two tetrahydrofuran rings (rings B and D) and one nonatomic ring C. Finally, the presence of a butyryloxy group at C-3 was deduced by the diagnostic HMBC correlations from H
2-2′ to C-1′ and from H-2 to C-1′. In addition, the key HMBC correlations from H-8 to C-1″ and from H
3-2″ to C-1″ indicated the connection of an acetyloxy group at C-8. Thus, the structure of
1 was determined as drawn in
Figure 2, with an uncommon tetracyclic ring system.
The relative configuration of
1 was determined by a detailed analysis of its NOESY spectrum (
Figure S6). As show in
Figure 2, the NOE correlations of H-1/H-10, H-1/H-13, H-8/H-10, and H-8/H
3-16 suggested that H-1, H-8, H-10, H-13, and H
3-16 were all co-facial, arbitrarily assigned as
β-configuration. The opposite (
α) orientation of H-2, H-6, H-9, H-14, and H
3-15 was indicated by the NOE cross peaks of H-2/H-9, H-2/H-14, H-2/H
3-15, and H-6/H-9. Finally, the relative configuration of
1 was established as 1
R*,2
S*,3
R*,6
R*,7
R*,8
S*,9
S*,10
R*,13
R*, 14
R*.
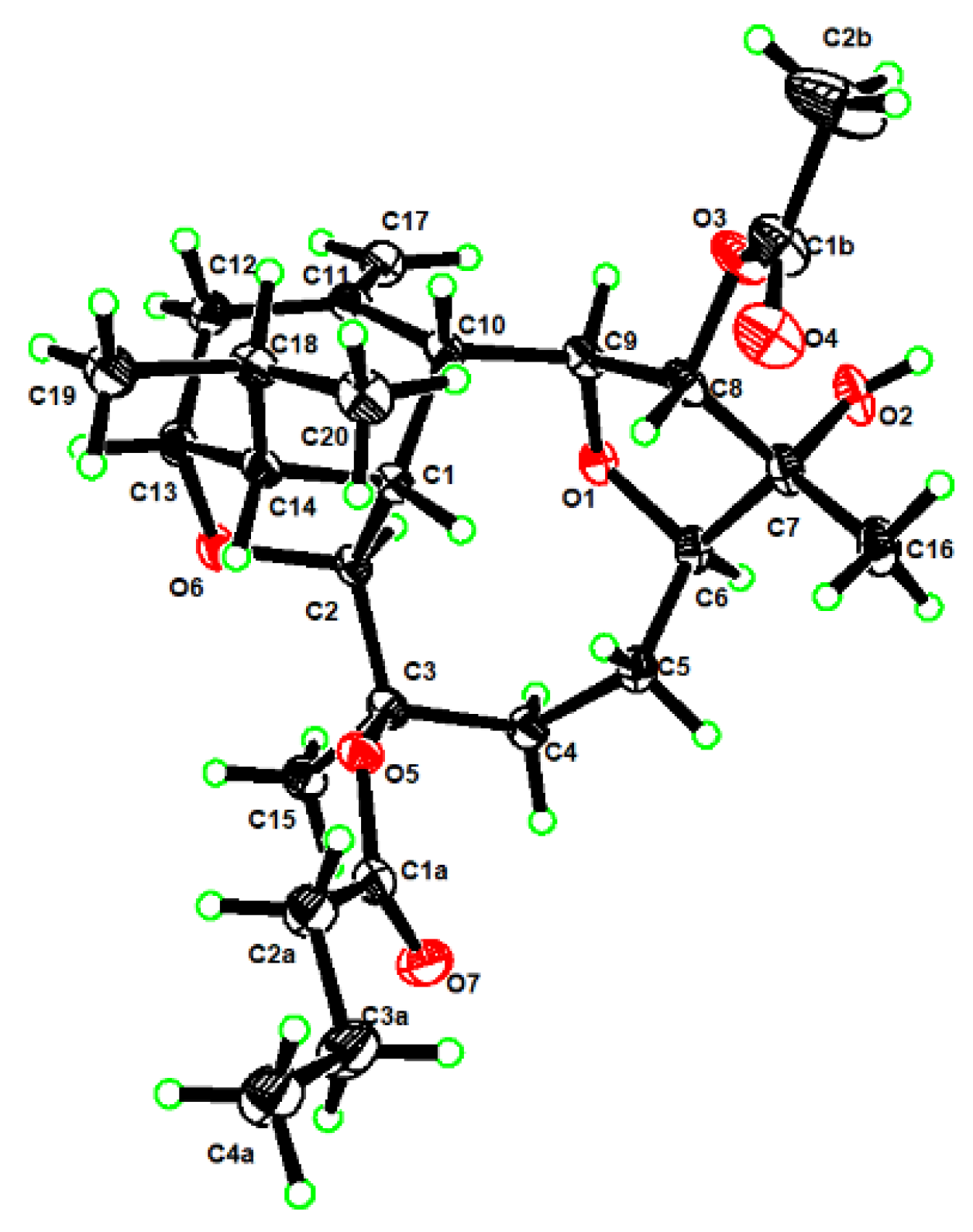
To determine the absolute configuration of
1, we fortunately managed to obtain its suitable single crystals in methanol, which were successful applied on X-ray crystallography using Cu K
α radiation (
λ = 1.54178 Å). The analysis of the X-ray data not only unambiguously confirmed the structure of
1 but also disclose its absolute configuration as 1
R,2
S,3
R,6
R,7
R,8
S,9
S, 10
R,13
R,14
R (Flack parameter was −0.09(7)) (
Figure 3, CCDC 2126980).
Compound
2 was isolated as colorless oil with the chemical formula of C
28H
44O
7 as disclosed by the HREIMS ion peak at
m/z 492.3080, ([M]
+, calcd 492.3082), implying seven degrees of unsaturation. The
13C NMR, DEPT and HSQC spectra (
Figures S10 and S11) revealed the presence of 28 carbons including six methyl groups, eight methylenes, nine methines, three quaternary carbons and two ester carbonyls (
δC 172.8, 172.9). In fact, compound
2 displayed very similar 1D NMR data as those of
1 (
Table 1), with the only difference on the substitution at C-8 position. Instead of the acetoxyl group in
1, the HMBC correlations (
Figure S12) from H-8 to 1″ and the
1H-
1H COSY correlations (
Figure S13) of H
2-2″ (
δH 2.38)/H
2-3″ (
δH 1.70)/H
3-4″ (
δH 0.98), indicating the butyryloxy group at the C-8 of
2, which was in agreement with a 28 mass units’ difference in their molecular weights. Therefore, the structure of
2 was determined as shown in
Figure 1, named ximaoornatin B.
Compound
3 was isolated as an optical active colorless oil. From the molecular ion peak at
m/z 422.2665 [M]
+ (calcd 422.2663) in the HRESIMS spectrum, a molecular formula of C
24H
38O
6 was elucidated, indicating six degrees of unsaturation. The 1D NMR data of
3 were reminiscent of those of
1 and
2 (
Table 1), and a further analysis of their 2D NMR spectra (
Figure 2,
Figures S19−S22) suggested the same skeleton of the three compounds with the same tetrahydrofuran rings A−D. The main differences between these compounds were found to be the presence of a hydroxy group at C-8 (
δC 79.0;
δH 4.09) in
3 instead of the acetoxyl group in
1 and butyroxyl group in
2, which was also confirmed by their mass spectrum. Thus, compound
3 was C-8 deacetyl derivative of
1, named ximaoornatins C.
The relative configurations of 2 and 3 were assigned to be the same as that of 1 due to the same NOE patterns in all three compounds. The absolute configurations of 2 and 3 were also assigned to be same as that of 1 by comparing their NMR spectra and on a biogenetic consideration since they only differed by the different substitution on 8-OH.
The molecular formula of compound
4 was found to be C
26H
40O
6 by HRESIMS (
m/z 471.2718 [M + Na]
+, calcd 471.2717), suggesting seven degrees of unsaturation. The
13C NMR and HSQC spectra (
Figures S26 and S27) disclosed the presence of 26 carbons including six sp
2 carbon atoms (2 × C=O, 1 × C = CH
2, 1 × C = CH) at lower field and twenty sp
3 carbon atoms at higher field (4 × OCH, 1 × OC, 5 × CH
2, 4 × CH, 6 × CH
3), accounting for four degrees of unsaturation. Thus, the remaining three degrees of unsaturation reveal
4 as a tricyclic molecule. Detailed analysis of its NMR data indicated that the spectroscopic features of
4 were similar to those of the known compound litophynin B (
6) [
11]. The apparent downfield shift of H-13 (from
δH 1.75, 1.05 in
6 to
δH 3.57 in
4) and C-13 (from
δC 25.4 in
6 to
δC 72.3 in
4) indicated the presence of 13-OH in
4, which was implied by the clear COSY correlations (
Figure S29) of H
2-12 (
δH 2.46, 2.31)/H-13/H-14 (
δH 1.40). In addition, the significant HMBC cross-peaks (
Figure S28) from H-8 (
δH 4.83) to C-1″ (
δC 170.6)/C-2″ (
δC 21.4), and from H-2″ to C-1″ confirmed the replacement of a butyryloxy group at C-8 in litophynin B by an acetyloxy group in
4. Therefore, the structure of
4 was defined as shown in
Figure 1.
The relative configuration of
4 was confirmed by NOESY experiment (
Figure 2 and
Figure S30). The NOE correlations (
Figure 2) of H-5/H
3-16 proved that the Δ
6,7 double bond has
E-geometry. The NOE correlations between H-1 (
δH 2.03) and H-10 (
δH 2.82), H-1 and H-13, and H-8 and H-10, suggested that H-1, H-8, H-10, and H-13 were
β-oriented. The correlations of H-2 (
δH 3.86)/H-14, H-2/H
3-15 (
δH 1.47), and H-9/H-14, suggested that of all of H-2, H-9 (
δH 4.09), H-14, and H
3-15 are
α-oriented. From the above evidence, the relative configuration of
4 was determined as 1
R*,2
R*,3
R*,8
R*,9
S*,10
R*,13
R*,14
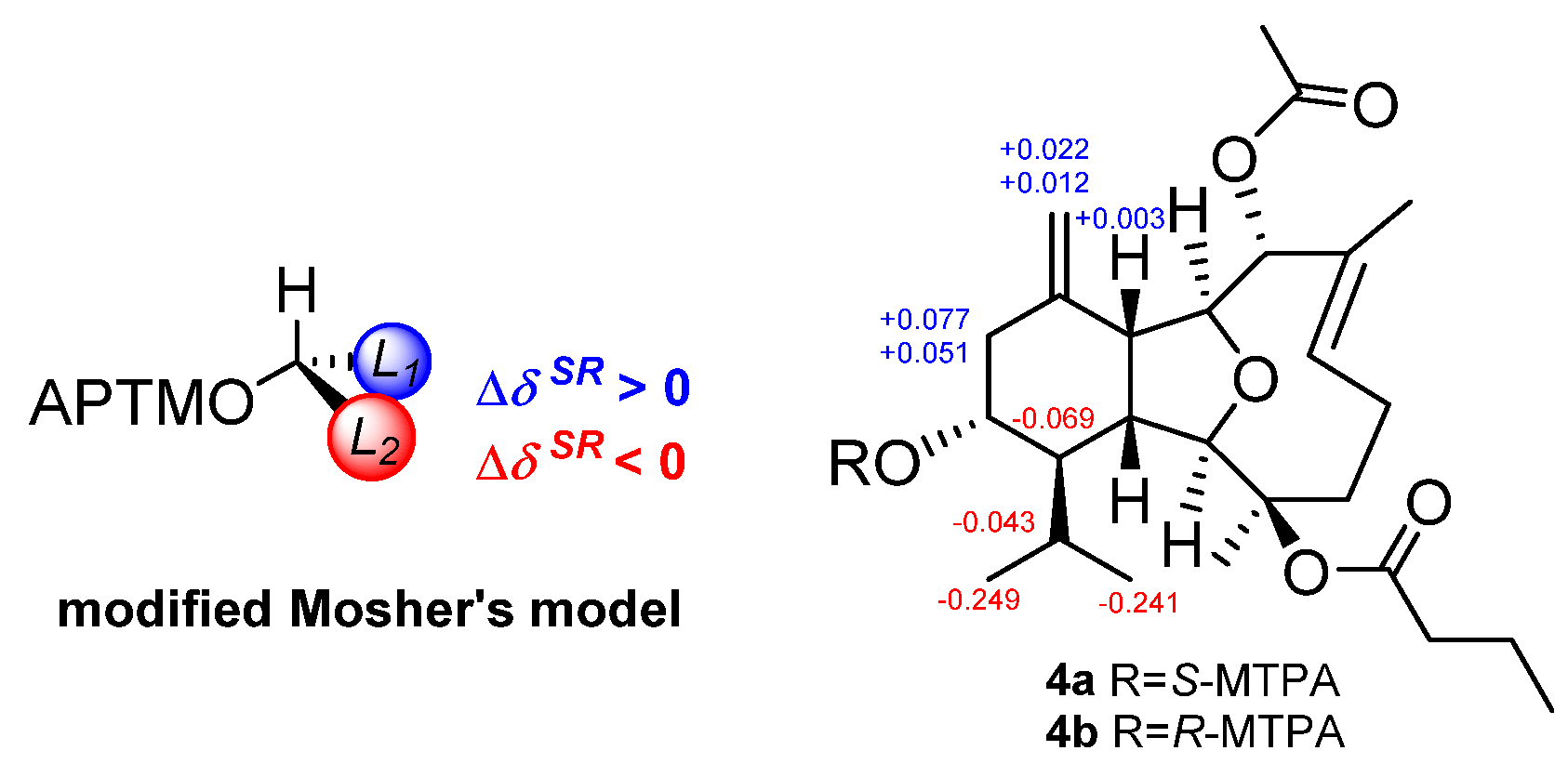
R*. Finally, to deduce the absolute configuration of the secondary alcohol at C-13, two aliquots of compound
4 were treated with (
R)- and (
S)-
α-methoxy-
α-trifluoromethylphenyl acetyl (MTPA) chlorides to obtain the (
S)- and (
R)-esters, respectively. The analysis of Δ
δSR values (
δS−
δR) observed for the signals of the protons close to 13-OH and according to Mosher’s rule [
12,
13], the absolute configuration at C-13 in
4 was established as
R (
Figure 4). Thus, the stereochemistry of
4 was unambiguously elucidated as 1
R,2
R,3
R,8
R,9
S,10
R,13
R,14
R.
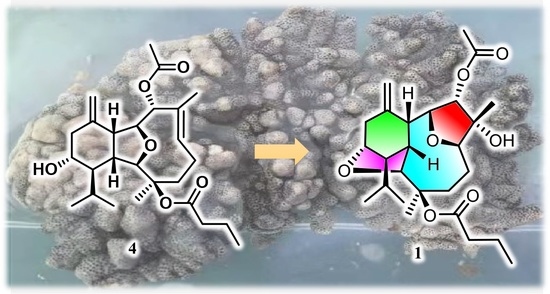
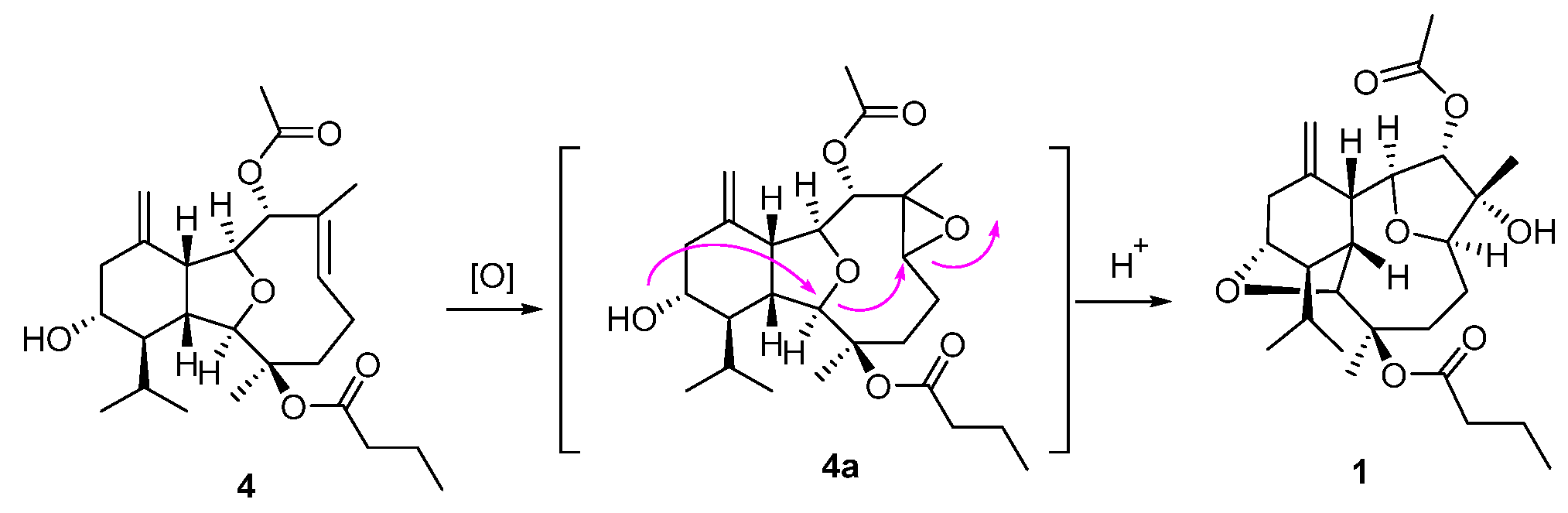
Compounds
1–
3 comprise an unprecedented tetradecahydro-2,13:6,9-diepoxybenzo[10]annulene skeleton, which were totally different from the co-occurring compound
4 and other eunicellin-type diterpenes. However, they are structurally related by sharing some common moieties, such as a six-membered ring A. Therefore, a plausible biosynthetic connection from the eunicellin diterpene
4 to
1 was proposed as drawn in
Scheme 1, which mainly underwent an oxidation of Δ
6,7 on
4 towards the epoxide intermediate
4a, followed by an acid-promoted electron delivery from the 13-hydroxyl to 6,7-epoxyl via the 2,9-ether group. All the isolates were subjected to the test of anti-inflammatory effects on LPS-induced TNF-α release in RAW264.7 macrophages and PTP1B inhibitory effects, and none of them showed obvious activities.
3. Materials and Methods
3.1. General Experimental Procedures
IR spectra were recorded on a Nicolet 6700 spectrometer (Thermo Scientific, Waltham, MA, USA); peaks are reported in cm−1. Melting points were measured on an X-4 digital micro-melting point apparatus. Optical rotations were measured on a Perkin-Elmer 241MC polarimeter (PerkinElmer, Fremont, CA, USA). The NMR spectra were measured at 300 K on DRX 500 and Avance 600 MHz NMR spectrometers (Bruker Biospin AG, Fallanden, Germany). Chemical shifts are reported in parts per million (δ) in CDCl3 (δH reported referred to CHCl3 at 7.26 ppm; δC reported referred to CDCl3 at 77.16 ppm) and coupling constants (J) in Hz; assignments were supported by 1H–1H COSY, HSQC, HMBC, and NOESY experiments. HR-ESIMS was carried out on a Waters Q-TOF Ultima mass spectrometer (Waters, MA, USA). HREIMS spectra were carried out on a Thermo DFS mass spectrometer. Semi-preparative HPLC was performed on an Agilent-1260 system equipped with a DAD G1315D detector using ODS-HG-5 (250 mm × 9.4 mm, 5 µm) by eluting with the CH3OH–H2O or CH3CN–H2O system at 3.0 mL/min. Commercial silica gel (200−300 and 300−400 mesh; Qingdao, China) was used for column chromatography (CC). Precoated Si gel plates (Merck Chemicals Co., Ltd., G60 F254, Shanghai, China) were used for analytical TLC. Sephadex LH-20 (Amersham Biosciences, London, U.K.) was also used for CC. All solvents used for column chromatography and HPLC were of analytical grade (Shanghai Chemical Reagents Co., Ltd., Shanghai, China) and chromatographic grade (Dikma Technologies Inc., Shanghai, China), respectively. X-ray diffraction studies were carried out on a Bruker D8 Venture diffractometer with Cu Kα radiation (λ = 1.54178 Å).
3.2. Biological Materials
Specimens of the soft coral Sinularia ornata, identified by Prof. Xiu-Bao Li from Hainan university, were collected along the coast of Ximao Island, Hainan province, China, in 2018, and were frozen immediately after collection. A voucher specimen (18-XD-07) was deposited at the Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China.
3.3. Extraction and Isolation
The frozen materials (943 g, dry weight) were cut into pieces and exhaustively extracted with Me2CO at room temperature. The organic extract was evaporated to give a brown residue, which was partitioned between Et2O and H2O. The Et2O solution was concentrated under reduced pressure to give a dark brown residue (36.3 g), which was fractionated by gradient Si gel (200−300 mesh) column chromatography (CC) (0 → 100% Et2O in petroleum ether (PE), yielding eight fractions (A−F). Fraction D was isolated by Sephadex LH-20 (PE/CH2Cl2/MeOH, 2:1:1), followed by silica gel CC (PE/CH2Cl2, 10:0 → 0:10) to give two subfractions (D2E and D2G). Fraction D2E was finally purified by reversed-phase HPLC (MeCN/H2O, 70:30; 3.0 mL/min) to give compound 1 (11.6 mg, tR = 17.1 min) and 4 (12.3 mg, tR = 12.3 min), while compound 2 (1.2 mg, tR = 18.6 min) was isolated from subfraction D2G by RP-HPLC (MeCN/H2O, 60:40; 3.0 mL/min). Fraction E was fractioned by Sephadex LH-20 (PE/CH2Cl2/MeOH, 2:1:1), followed by silica gel CC (PE/Et2O, 10:0 → 0:10) to give two subfractions (E3E and E3H). Subfraction E3E was further purified by stepwise HPLC (MeCN/H2O, 62:38 → 70:30; 3.0 mL/min) to obtain compound 5 (17.0 mg, tR = 5.3 min). Similarly, compound 3 (2.0 mg, tR = 13.5 min) was isolated from fraction E3H by reversed-phase HPLC (MeCN/H2O, 75:25 → 98:2; 3.0 mL/min).
Ximaoornatin A (
1): colorless crystals; m.p. 166.2~166.4 °C; [
α]
−46.7 (
c 0.35, CHCl
3); IR (KBr)
νmax = 3445, 2919, 2849, 1959, 1620, 1384, 1156, 1043 cm
−1;
1H and
13C NMR data see
Table 1; HR-ESIMS
m/z 487.2670 [M + Na]
+ (calcd. for C
26H
40NaO
7, 487.2666).
Ximaoornatin B (
2): colorless oil; [
α]
−15.4 (
c 0.12, CHCl
3); IR (KBr)
νmax = 3441, 2960, 2924, 2870, 1959, 1732, 1620, 1384, 1260, 1074, 1040 cm
−1;
1H and
13C NMR data see
Table 1; HR-EIMS
m/z 492.3080 [M]
+ (calcd. for C
28H
44O
7, 492.3082).
Ximaoornatin C (
3): colorless oil; [
α]
−55.0 (
c 0.11, CH
3OH); IR (KBr)
νmax = 3444, 2924, 1959, 1731, 1620, 1384, 1260, 1045, 800 cm
−1;
1H and
13C NMR data see
Table 1; HR-EIMS
m/z 422.2665 [M]
+ (calcd. for C
24H
38O
6, 422.2663).
Litophynin K (
4): colorless oil; [
α]
−58.4 (
c 0.24, CHCl
3); IR (KBr)
νmax = 3446, 2925, 1959, 1733, 1665, 1384, 1247, 1097, 1051 cm
−1;
1H and
13C NMR data see
Table 1; HR-ESIMS
m/z 471.2718 [M + Na]
+ (calcd. for C
26H
40NaO
6, 471.2717).
X-ray Crystallographic Analysis for
1. Colorless blocks, C
2.08H
3.2O
0.56,
Mr = 37.17, monoclinic, crystal size 0.15 × 0.08 × 0.05 mm
3, space group
P2
1,
a = 11.0532(5) Å,
b = 9.4225(4) Å,
c = 12.3134(5) Å,
V = 1271.54 (9) Å
3,
Z = 25,
Dcalcd = 1.213 g/cm
3,
F(000) = 504.0, 19,814 reflections measured (7.24° ≤ 2Θ ≤ 149.14°), 5096 unique (
Rint = 0.0470,
Rsigma = 0.0381) which were used in all calculations. The final
R1 was 0.0346 (
I > 2
σ(
I)) and w
R2 was 0.0901 (all data). The X-ray measurements were made on a Bruker D8 Venture X-ray diffractometer with Cu K
α radiation (
λ = 1.54178 Å) at 170.0 K. The structure was solved with the ShelXT structure solution program using Intrinsic Phasing and refined with the ShelXL refinement package using least squares minimization. Crystallographic data for
1 were deposited at the Cambridge Crystallographic Data Centre (Deposition nos. CCDC 2126980). Copies of these data can be obtained free of charge via
www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB21EZ, UK (fax: +44-1223-336-033; e-mail:
deposit@ccdc.cam.ac.uk).
3.4. Anti-Inflammatory Activity Assay
RAW264.7 cell, a murine macrophage cell line, was obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). In the bioassay for anti-inflammation, RAW264.7 cells were grown in DMEM containing 2 mmol/L L-glutamine, 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin, and maintained in a humidified incubator of 5% CO2 at 37 °C. The anti-inflammatory effect was measured by the cell viability and TNF-α production of RAW264.7 cells. The cells (1 × 105/well) were incubated in 96-well plates in triplicate. For the cell viability part, RAW264.7 cells were cultured with vehicle (final concentration of 0.125% DMSO) or tested compounds at the indicated concentrations for 24 h. A total of 20 μL CCK-8 reagent was added to each well and after 1 h incubation and the OD values were collected after 1 h incubation at 450 nm (650 nm calibration) by a microplate reader (Molecular Devices, Sunnyvale, CA, USA). For the anti-inflammatory activity assay, after adherence, the cells were cultured with vehicle (final concentration of 0.125% DMSO) or tested compounds at the indicated concentrations for 30 min. Then, the cells were primed with 1 μg/mL of LPS (Lipopolysaccharide) for 24 h. Supernatants were centrifuged and then quantified with the mouse TNF-α ELISA kit following the manufacturer’s instructions. The CC50 and IC50 were estimated using the log (inhibitor) vs. normalized response nonlinear fit (Graph Pad Prism 6.0).
3.5. PTP1B Inhibitory Activity Assay
The recombinant PTP1B catalytic domain was expressed and purified according to a previous report [
13]. The enzymatic activities of the PTP1B catalytic domain were determined at 30 °C by monitoring the hydrolysis of
pNPP. The dephosphorylation of
pNPP generates product
pNP, which was monitored at an absorbance of 405 nm by the EnVision multilabel plate reader (PerkinElmer Life Sciences, Boston, MA, USA). In a typical 100 μL assay mixture containing 50 mmol/L 3-[N-morpholino]-propanesulfonic acid (MOPs), pH 6.5, 2 mmol/L
pNPP, and 30 nmol/L recombinant PTP1B, activities were continuously monitored and the initial rate of the hydrolysis was determined using the early linear region of the enzymatic reaction kinetic curve. The IC50 was calculated with Prism 4 software (Graphpad, San Diego, CA, USA) from the nonlinear curve fitting of the percentage of inhibition (% inhibition) vs. the inhibitor concentration [I] using the following equation: % inhibition = 100/(1 + [IC50/[I]]
k), where
k is the Hill coefficient.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}