1. Introduction
Mucormycosis, also called zygomycosis and black fungus, is a rare, non-contagious fungal infection that can cause high morbidity and mortality. The mucormycetes are a notorious group of fungi belonging to the Mucorales order and Mucoromycotina subphylum [
1]. These molds can commonly be found in the soil, on plant surfaces, and in decaying fruits, vegetables, bread, and other compost piles. A majority of mold species are harmless as they are incapable of tolerating human body temperature. Unfortunately, a thermotolerant type was recently identified in COVID-19 patients worldwide [
2], especially in India. An individual can be infected by inhaling or through burns and open wounds on the skin coming into close contact with the spores [
3]. Upon infection, the host’s blood vessels are blocked as the spores produce hyphae, which lead to swelling and eventual death of the surrounding tissues. This infection can affect the brain, lungs, skin, and sinuses [
4,
5], with the severity of the outcome depending on the immune system’s efficacy and certain underlying medical conditions. Certain medical conditions, such as diabetes mellitus, various types of cancers, organ or stem cell transplants, neutropenia, hemochromatosis, metabolic acidosis, and viral diseases such as HIV, and recently, SARS-CoV-2 [
6], are known to enhance the risk of mucormycosis infection.
The mortality rate of mucormycosis before COVID-19 was around 54% [
7], but disseminated mucormycosis caused 96% fatality [
8]. The combination of SARS-CoV-2 and disseminated mucormycosis is, therefore, the worst-case scenario for any individual [
9]. This mold infection is considered to be extremely opportunistic, causing severe damage to the host. India is the country most impacted by this sudden epidemic. Over 47,000 cases were reported in India by July 2021. Out of which, approximately 28,000 were active cases [
10,
11]. For the period of August 2021 to February 2022, the exact number of COVID-19-associated mucormycosis cases in India remains uncertain. However, besides India, at least 17 other countries reported CAM cases. These countries were the United States of America, Mexico, France, Germany, Austria, the Netherlands, United Kingdom, Czech Republic, Italy, Kuwait, Iran, Turkey, Lebanon, Brazil, Chile, China, Pakistan, and Bangladesh [
12].
In particular, there is an uptrend in orbital mucormycosis cases post COVID-19 infection. The COVID-19-associated mucormycosis (CAM) primarily affects the sinus and the bony cavities holding the eyeballs. There is a connection between black fungus infection and the usage of steroids to treat COVID-19 patients. The steroids are used to reduce inflammation in the lungs of the patients but also cause blood sugar levels to rise abnormally, making an individual prone to mucormycosis infection [
13]. CAM was found to be prevalent in the male population (78.9%). Cases that emerged due to diabetes mellitus and corticosteroid were reported to be 80% and 76.3%, respectively. Around 30.7% of CAM patients died [
14]. Current medical treatment options include amphotericin B, isavuconazole, posaconazole, and invasive treatment for surgical removal of infected tissues [
15]. These antifungal drugs and surgeries have serious side effects and limited efficacy. Therefore, safe alternatives to treat mucormycosis are urgently needed.
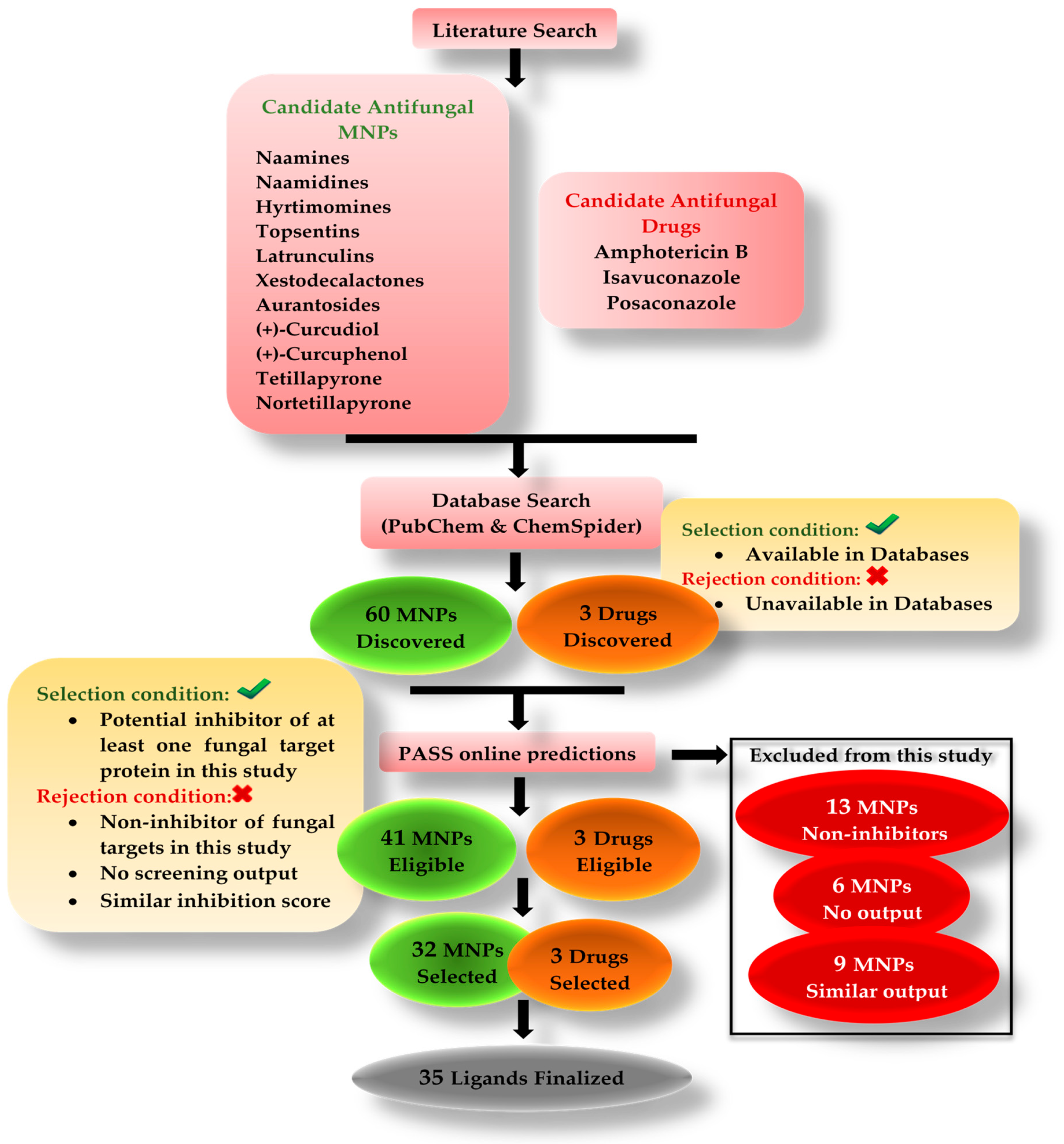
Many studies are being conducted around the globe testing natural bioactive compounds to treat SARS-CoV-2 and mucormycosis. Bioactive compounds from marine organisms hold the potential to inhibit different viruses and fungi [
16,
17,
18,
19,
20,
21,
22]. The purpose of this in silico study was to investigate the antifungal potential of bioactive compounds originating from marine sponges. Our research included six different classes of compounds such as alkaloids (naamines, naamidines [
22,
23,
24,
25,
26], hyrtimomines [
27,
28], and topsentins [
29,
30,
31,
32]), macrolides (latrunculins [
33,
34,
35,
36]), bioactive metabolites (xestodecalactones [
37,
38,
39]), sesquiterpene phenols [(+)-curcudiol, (+)-curcuphenol] [
40,
41,
42,
43], hydroxpyran-2-ones (tetillapyrone and nortetillapyrone [
44,
45,
46,
47,
48]), and tetratomic acid glycosides (aurantosides [
49,
50,
51,
52,
53]). These compounds were probed for seven therapeutic targets belonging to
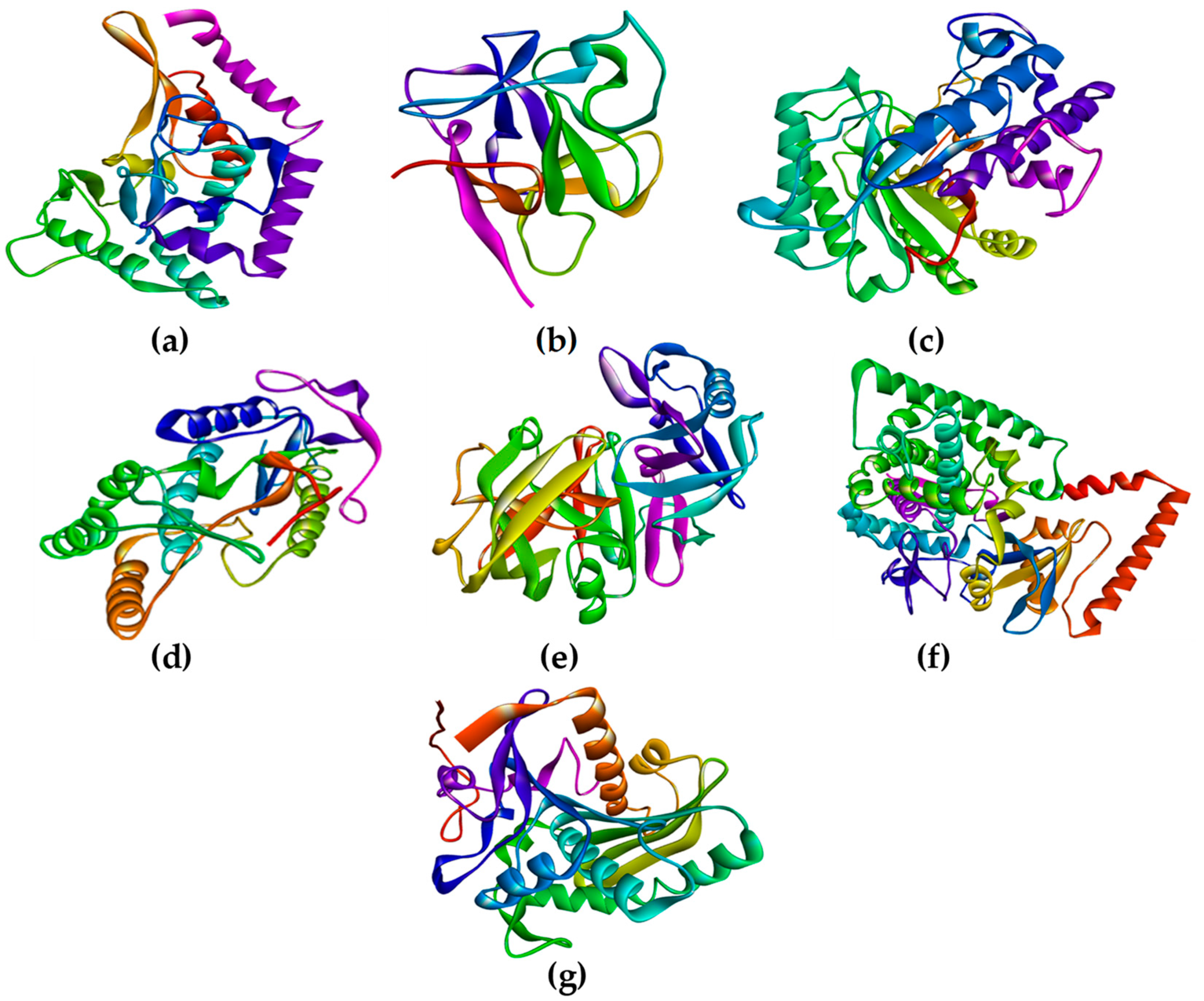
Rhizopus delemar,
Candida albicans,
Rhizopus arrhizus,
Rhizopus chinensis, and finally,
Rhizopus microsporus var. chinensis. These targets were CotH3, mucoricin, lanosterol 14 alpha-demethylase, exo-1,3-beta-glucan synthase, RdRp, fungal lipase, and rhizopuspepsin, respectively. Three currently used drugs, amphotericin B, isavuconazole, and posaconazole were included to compare with selected antifungal candidates. This research pinpointed the suitable fungal targets and highlighted specific MNPs based on their potential targeted biological activity. Ultimately, the drug-likeness properties, as well as the potentially harmful and toxic properties of the MNPs above, were evaluated and elucidated in this work.
3. Discussion
SARS-CoV-2 mortality and cases are much higher than originally thought [
60]. This virus has shown flexibility in evolutionary terms and most likely may continue to exist for several years, in its current form or even in an evolved form [
61]. As long as SARS-CoV-2 exists, the risk of mucormycosis infections might remain a subject of major concern. The world is now desperately looking for relief from the SARS-CoV-2 pandemic and mucormycosis epidemic. This paper proposes the idea of considering marine-sponge-based antifungal compounds against mucormycosis.
According to the PASS online program, some compounds belonging to the naamine group were antifungal, anti-asthmatic, anti-infective, antiviral, and can be used for mucositis treatment. These features help deal with both viral and fungal infections. Naamines are also kinase inhibitors and histidine kinase inhibitors. This indicates that naamines can inhibit the target CotH3. Furthermore, they were found to inhibit the beta-glucuronidase enzyme. This makes them a potential inhibitor of mucoricin target. Moreover, they are capable of inhibiting rhizopuspepsin and the RdRp enzyme. Based on the molecular docking outcomes, naamine D had the highest binding affinity, −8.8 kcal/mol with RdRp. Molecular dynamic simulation in IMODS server also suggested that the RdRp-naamine D formed a stable complex. Drug-likeness analysis for naamines showed that they all followed Lipinski parameters, which makes them good drug candidates. Swiss-ADME outcome suggests that naamine A, B, E, and F are moderately soluble and only naamine D is poorly soluble. All naamines had a bioavailability of 0.55 and were highly absorbable by the GI tract. Naamine A, B, and D can permeate the blood-brain barrier. In addition, OSIRIS analysis showed that all naamines pose low to no risk of being an irritant, mutagenic agent, or tumorigenic agent, and they do not affect the reproductive system. ProTox-II indicated that naamines belonged to classes 4 and 5. PkCSM analysis labeled naamine A, B, F, G as being hepatotoxic. However, naamine D and E are not hepatotoxic. Naamines were not found to cause acute inhalation and dermal toxicity. However, they can cause acute oral toxicity. Naamine D does not cause skin sensitization, whereas other naamines may cause skin sensitization. All naamines can cause eye irritation and corrosion.
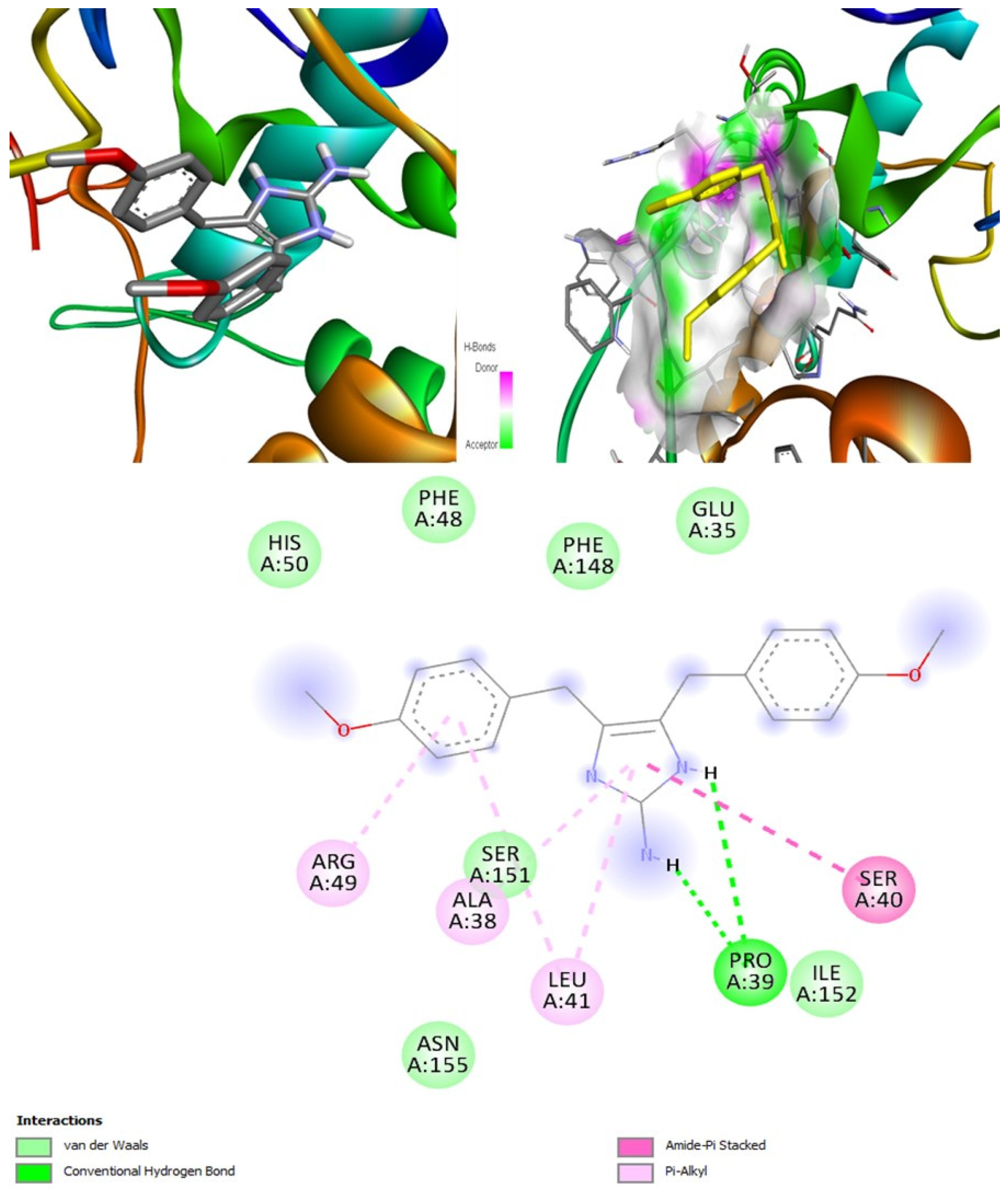
Naamidines A, B, and C are shown to possess antifungal, anti-asthmatic, anti-eczematic, and antiviral properties. In addition, they are kinase and histidine kinase inhibitors with good inhibition of CotH3 protein. Molecular docking analysis showed that naamidines overall had good binding affinities with some of the targets. Drug-likeness analysis for naamidines showed that they both followed Lipinski parameter, highlighting the potential to become a drug. Swiss-ADME outcome suggests that naamidine A, B, and C are poorly soluble. All naamidines had a bioavailability of 0.55 and are highly absorbable by the GI tract. None of them can permeate the blood-brain barrier. OSIRIS analysis showed that both naamidines pose a low risk of being an irritant, mutagenic agent, or tumorigenic agent, and they do not affect the reproductive system. PkCSM analysis suggests that naamidines A, B, and C are hepatotoxic. ProTox-II indicates that naamidines are class 5 compounds. Moreover, the StopTox outcome suggests that they do not cause acute inhalation or dermal toxicity, and there is no indication of skin sensitization. However, naamidines might cause eye irritation and corrosion. Among all of them, only naamidine C might exhibit oral toxicity.
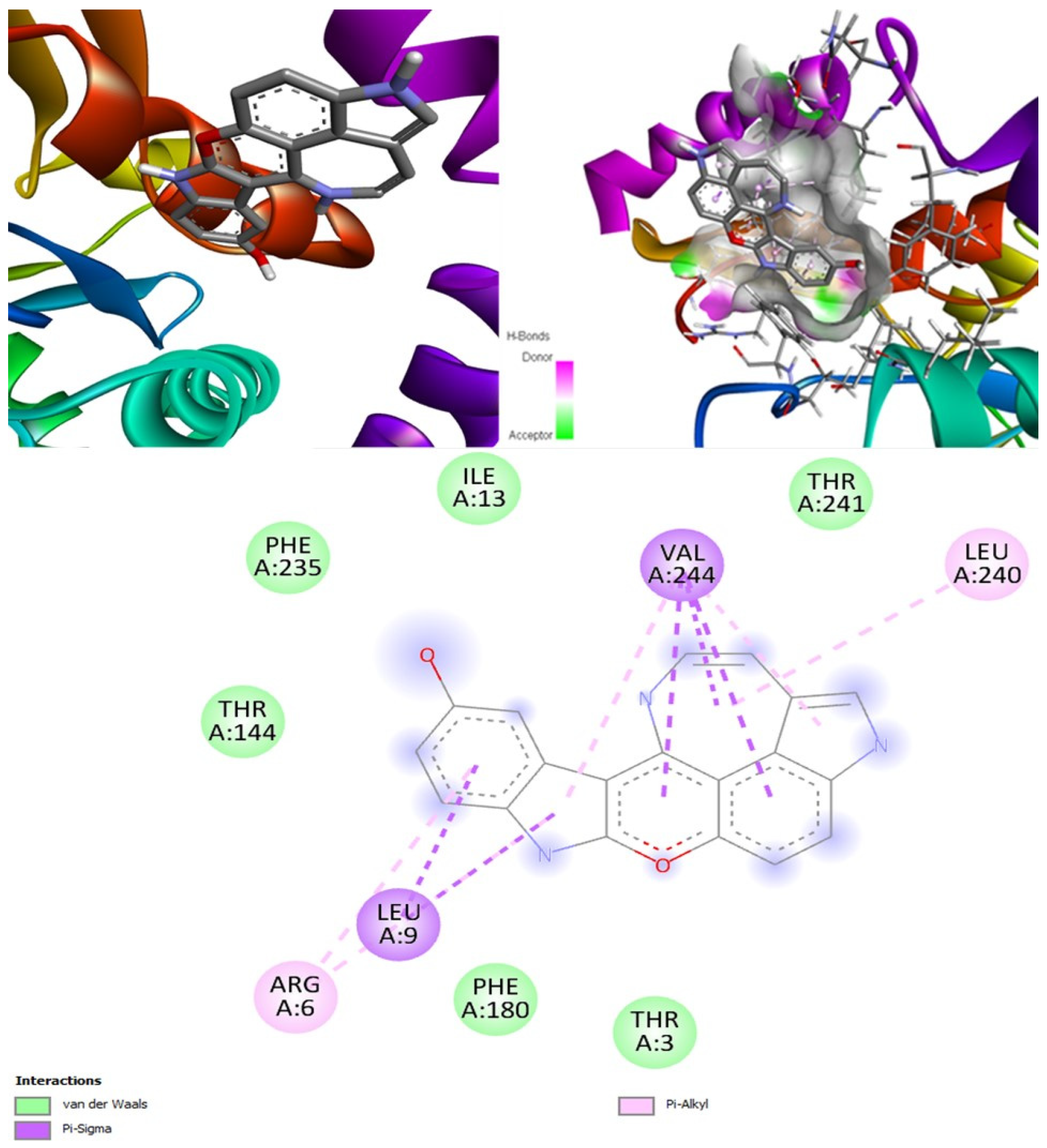
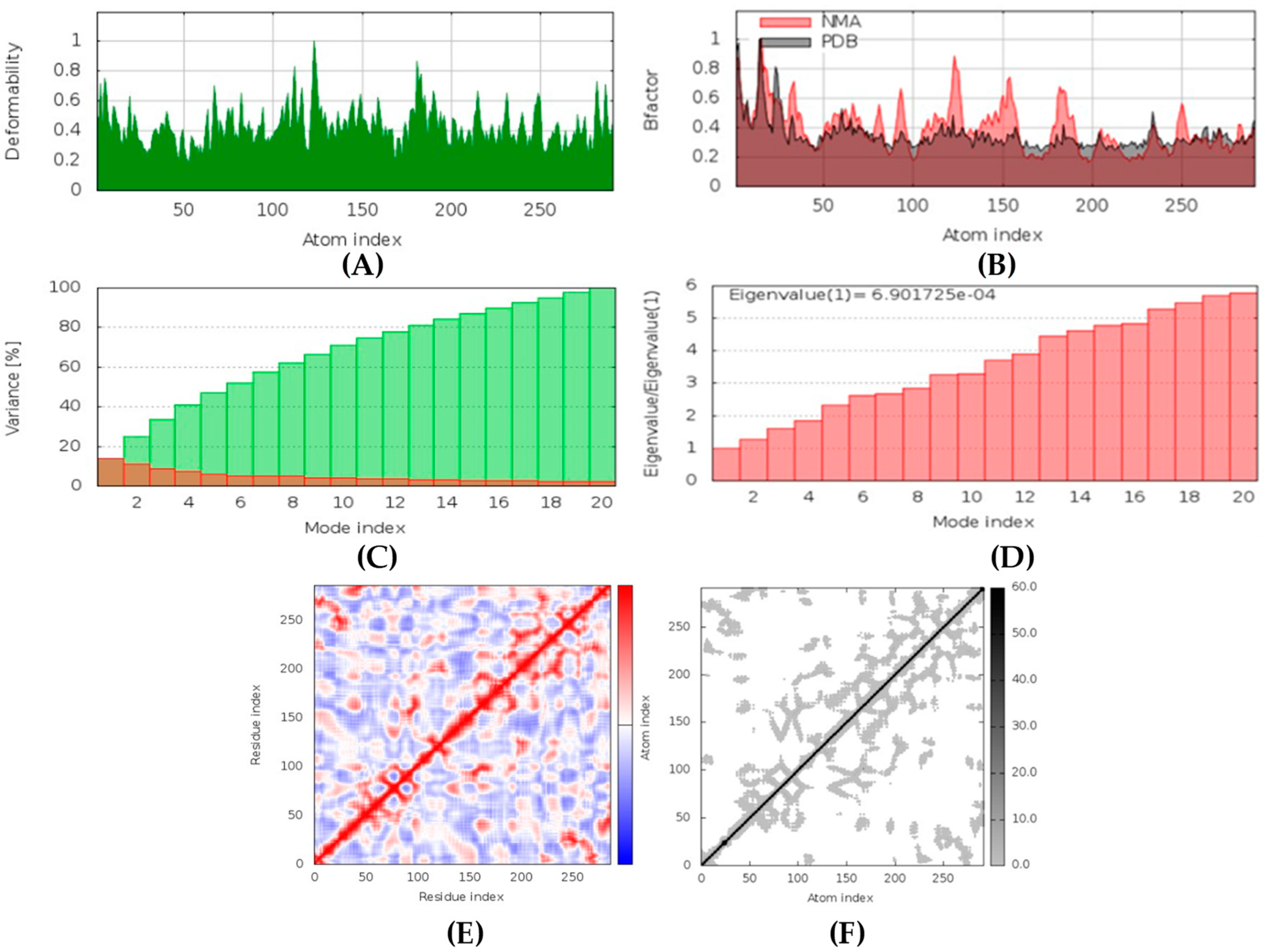
Hyrtimomines A, B, C, F, and G are potential antifungal, anti-infective, and anti-inflammatory agents that might help with COVID-19 and mucormycosis symptoms. They are strong inhibitors of kinase and histidine kinase (pa > 0.3 and pa > 0.7). In addition, they can also inhibit the beta-glucuronidase enzyme, indicating that they can inhibit both CotH3 and mucoricin proteins. The docking analysis showed that hyrtimomine A had the highest binding affinity, −9 kcal/mol, with CotH3 protein. MD simulation indicated that a stable complex was formed. Drug-likeness estimation suggests that hyrtimomines do not violate Lipinski’s rules. Thus, they are potential drug-like molecules. However, Swiss-ADME showed that they have poor solubility except hyrtimomine G. The bioavailability value was high, 0.55, and hyrtimomine B had 0.56. High GI absorption was indicated for hyrtimomine A, B, C, and F but not in the case of hyrtimomine G. No blood-brain barrier permeability was shown. OSIRIS analysis showed that hyrtimomines pose low risk of being an irritant or mutagenic agent, and they do not affect the reproductive system. However, hyrtimomine F might be tumorigenic. PkCSM analysis suggests that they are hepatotoxic. ProTox-II indicates that they belong to class 4, except hyrtimomine G, which is a class 3 MNP. StopTox found that they do not cause acute inhalation and dermal toxicity. Hyrtimomine C might be toxic in the case of oral exposure. Hyrtimomine B, C, F, and G might cause irritation and corrosion of eyes, while hyrtimomine A does not.
Topsentins were found to be antifungal and anti-infective. They are good inhibitors of kinase and histidine kinase which means they can inhibit CotH3 protein. Molinspiration analysis also confirmed the potential kinase inhibition potential of topsentins (
Table S22). Topsentins group showed good binding affinities in the molecular docking analysis. Drug-likeness estimation suggests that topsentins do not violate Lipinski rules and, therefore, they might meet necessary drug requirements. However, Swiss-ADME showed that they have poor solubility and bioavailability value was high, 0.55. High GI absorption indicated that topsentin D is blood-brain-barrier permeable. OSIRIS analysis showed that topsentins pose low risk of being an irritant, mutagenic agent, or tumorigenic agent, and they do not affect the reproductive system. PkCSM analysis suggests that only topsentin D is hepatotoxic. ProTox-II indicates that they belong to class 4. StopTox found that topsentins might not cause acute inhalation and dermal toxicity. However, topsentin A and D can cause oral toxicity. In addition, topsentin A and D are expected to be non-corrosive to the eye and skin.
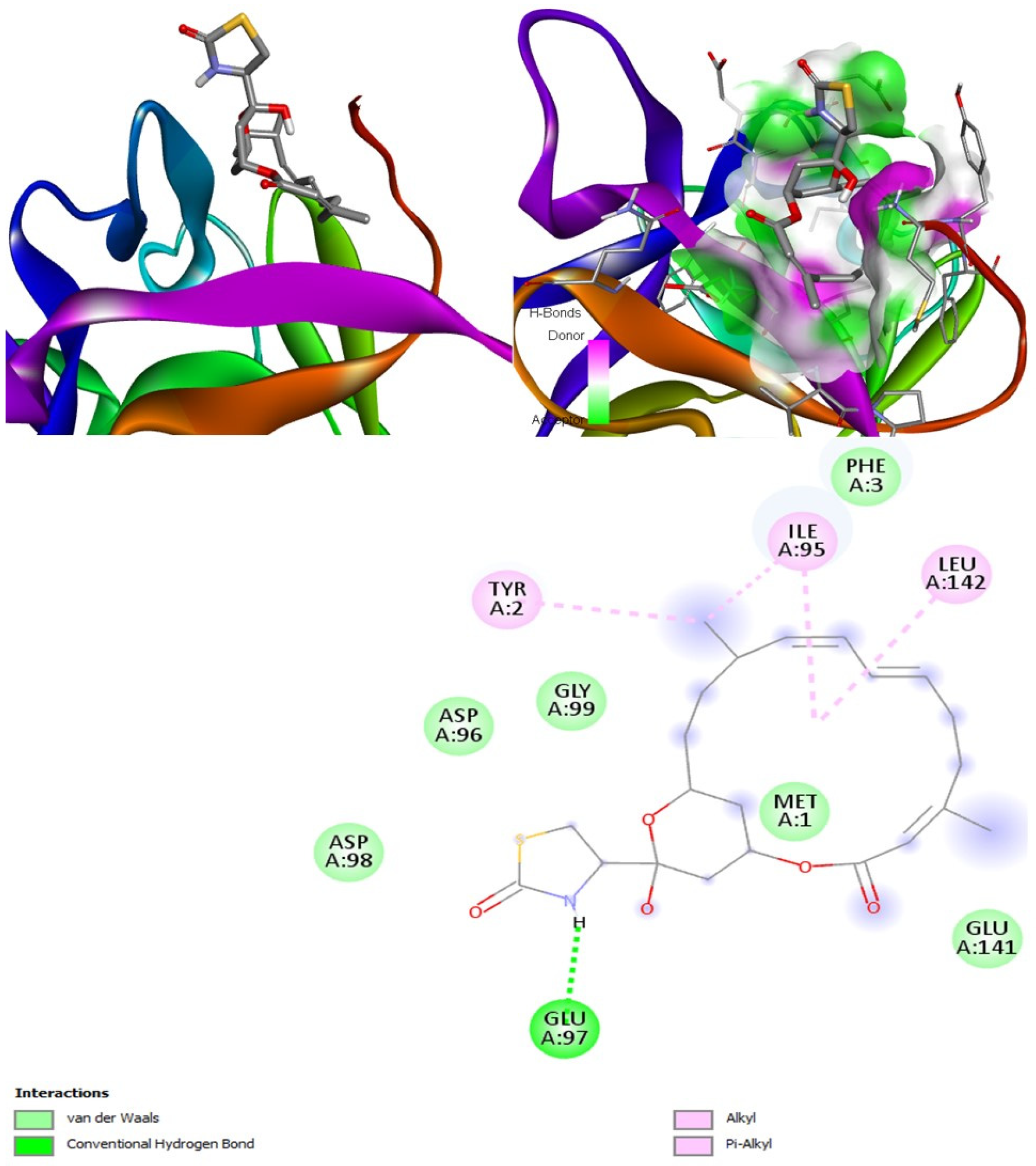
Latrunculin A, B, and S were found to be antifungal, anti-eczematic, antiviral, antibiotic, and antifungal enhancers. Apart from this, they can provide respiratory distress relief, making them a suitable candidate for COVID-19-associated mucormycosis. They are beta-glucuronidase and rhizopuspepsin inhibitors. Molecular docking showed that latrunculin S had the highest binding affinity of −9.8 kcal/mol with target rhizopuspepsin. Furthermore, latrunculin A had the highest binding affinity with the target mucoricin, −8.6 kcal/mol. MD simulations for both complexes were suggestive of a very stable conformation being formed. Drug-likeness estimation suggests that latrunculins do not violate Lipinski rules. Thus, they have drug-like properties. Swiss-ADME indicated that they have good solubility and bioavailability, 0.55. High GI absorption indicated that they are not able to cross the blood-brain barrier. OSIRIS analysis showed that latrunculins A, B, and S pose low to no risk of being an irritant, mutagenic agent, or tumorigenic agent, and they do not affect the reproductive system. PkCSM analysis suggests that latrunculin A, B, and S can be hepatotoxic. ProTox-II indicates that they belong to class 4. StopTox found that latrunculin should not cause acute inhalation or oral and dermal toxicity, but they can be corrosive to the eyes.
Xestodecalactones A, B, C, D, E, and F are predicted to be antifungal, anti-eczematic, antiviral, and anti-infective. In addition, they are kinase, histidine kinase, and beta-glucuronidase inhibitors; thus, they can inhibit CotH3 and mucoricin proteins. Molecular docking output for xestodecalactones was satisfactory. Drug-likeness estimation suggests that xestodecalactones do not violate Lipinski rules and are suitable to be called drug-like. Swiss-ADME analysis indicated that they have good solubility, except xestodecalactone E, which is moderately soluble. The bioavailability was 0.55, and GI absorption also indicated no potential to cross the blood-brain barrier. OSIRIS analysis showed xestodecalactones A, B, C, and D pose low to no risk of being an irritant, mutagenic agent, or tumorigenic agent, and they do not affect the reproductive system. However, E and F pose a high risk of being an irritant. PkCSM analysis suggests that xestodecalactones A, B, C, D, E, and F are non-hepatotoxic. ProTox-II indicates that the majority of them belong to class 4 and only xestodecalactone F is class 5. StopTox found that xestodecalactone A, B, and C should not cause skin sensitization, acute inhalation, and oral toxicity. However, xestodecalactone D might cause dermal toxicity. Xestodecalactone E might cause skin sensitization. Xestodecalactone F might cause both dermal toxicity and skin sensitization. Xestodecalactone A, B, and C can be corrosive to the eyes. On the other hand, D, E, and F should not cause eye and skin irritation and corrosion.
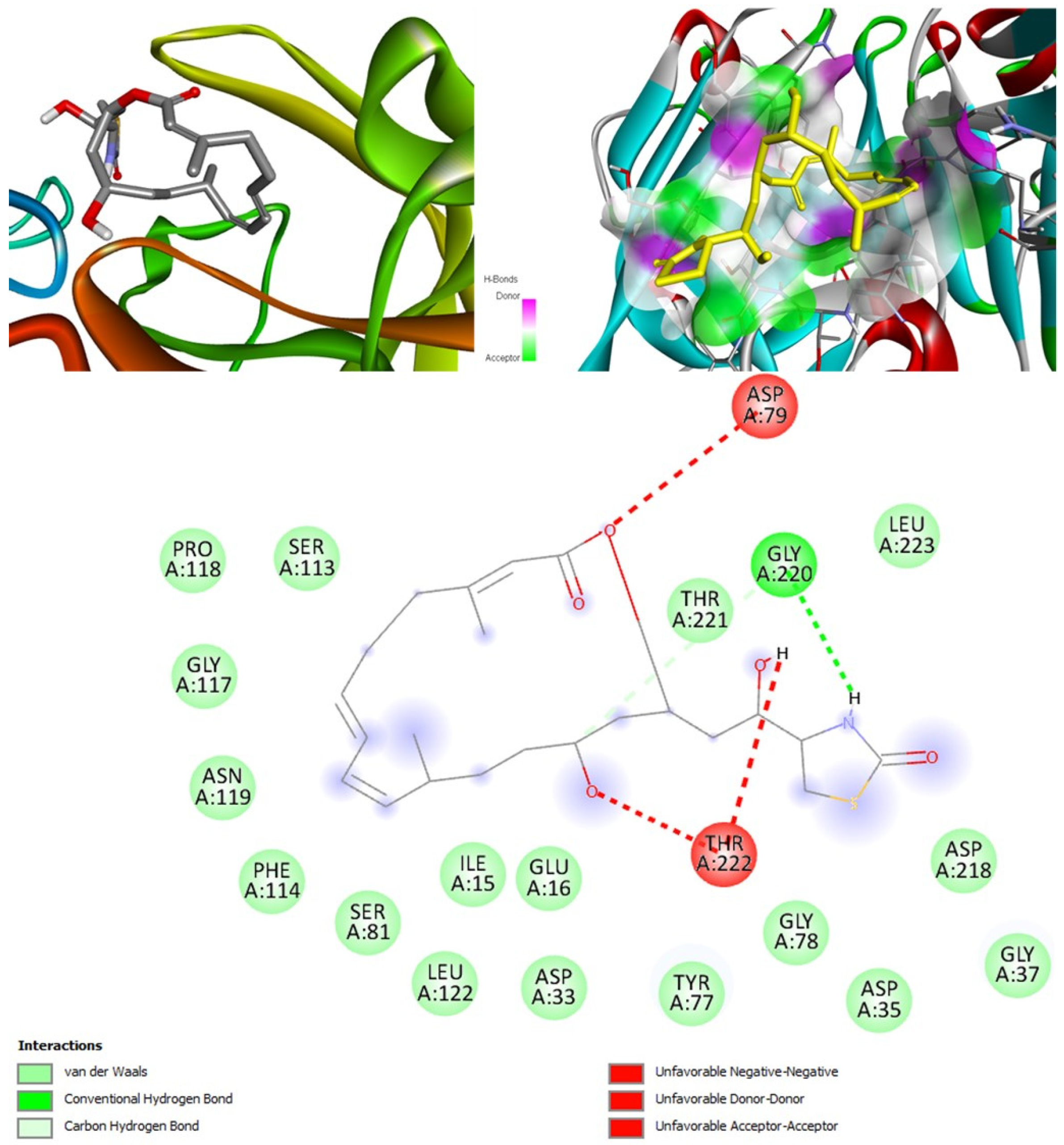
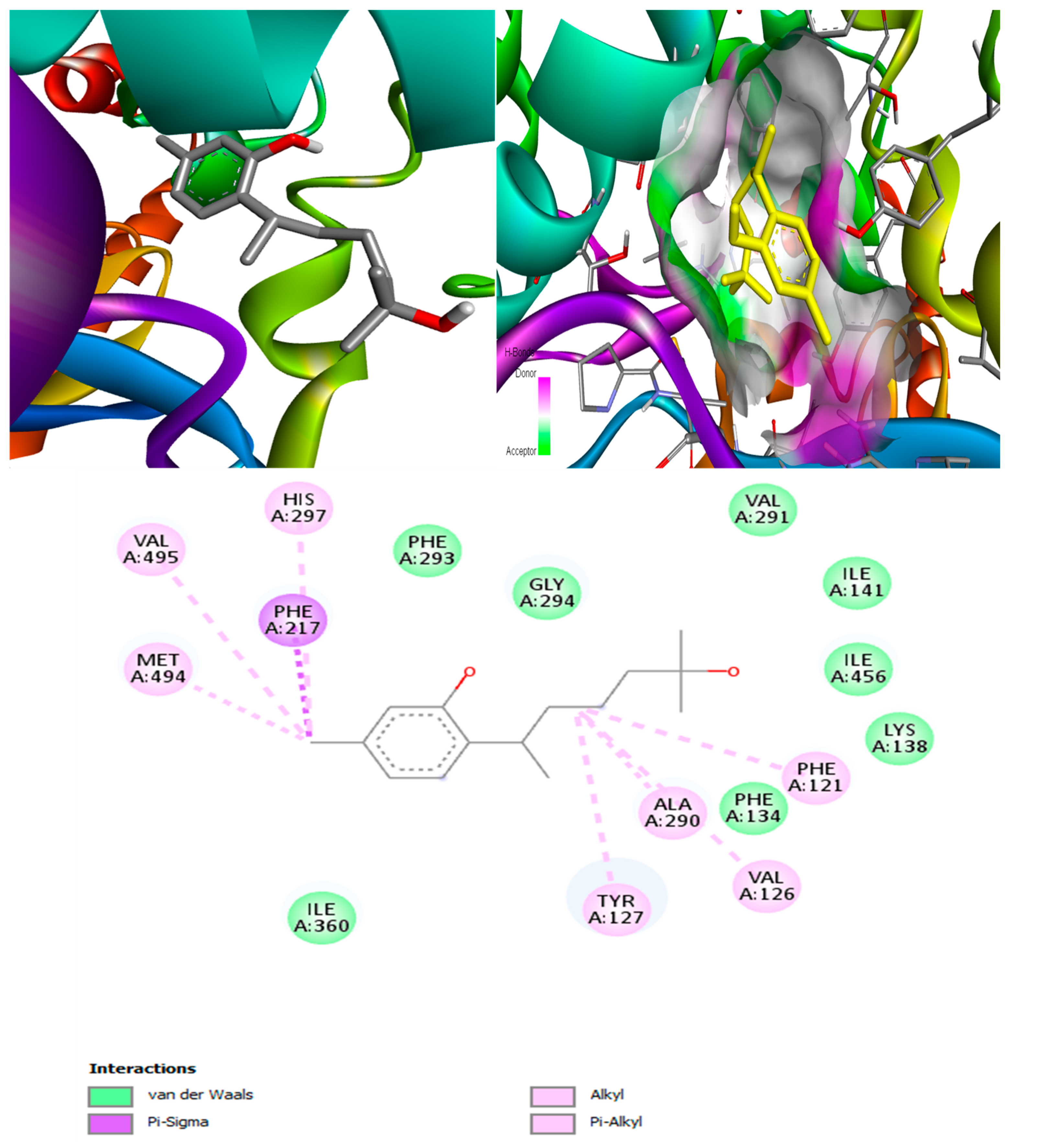
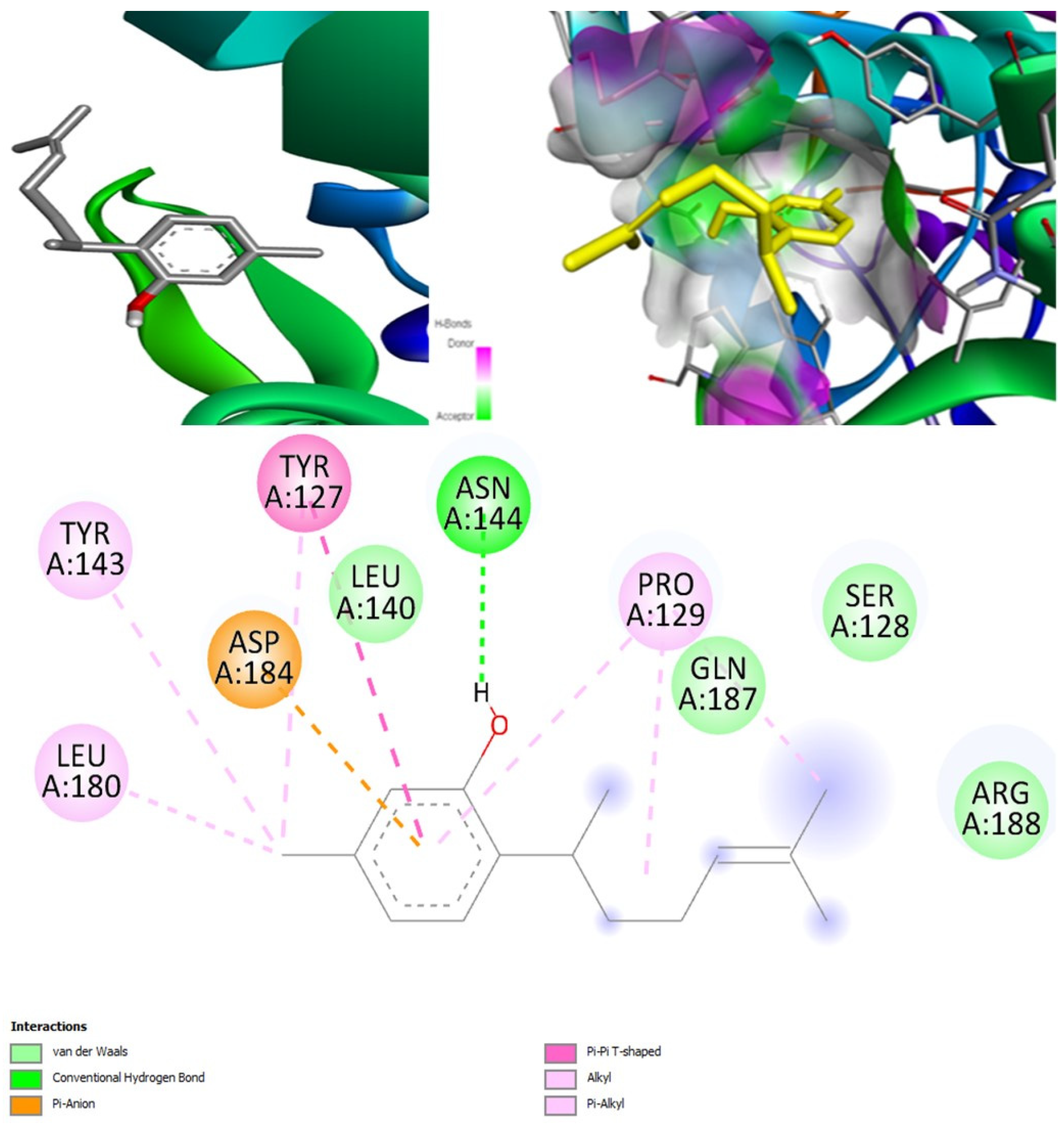
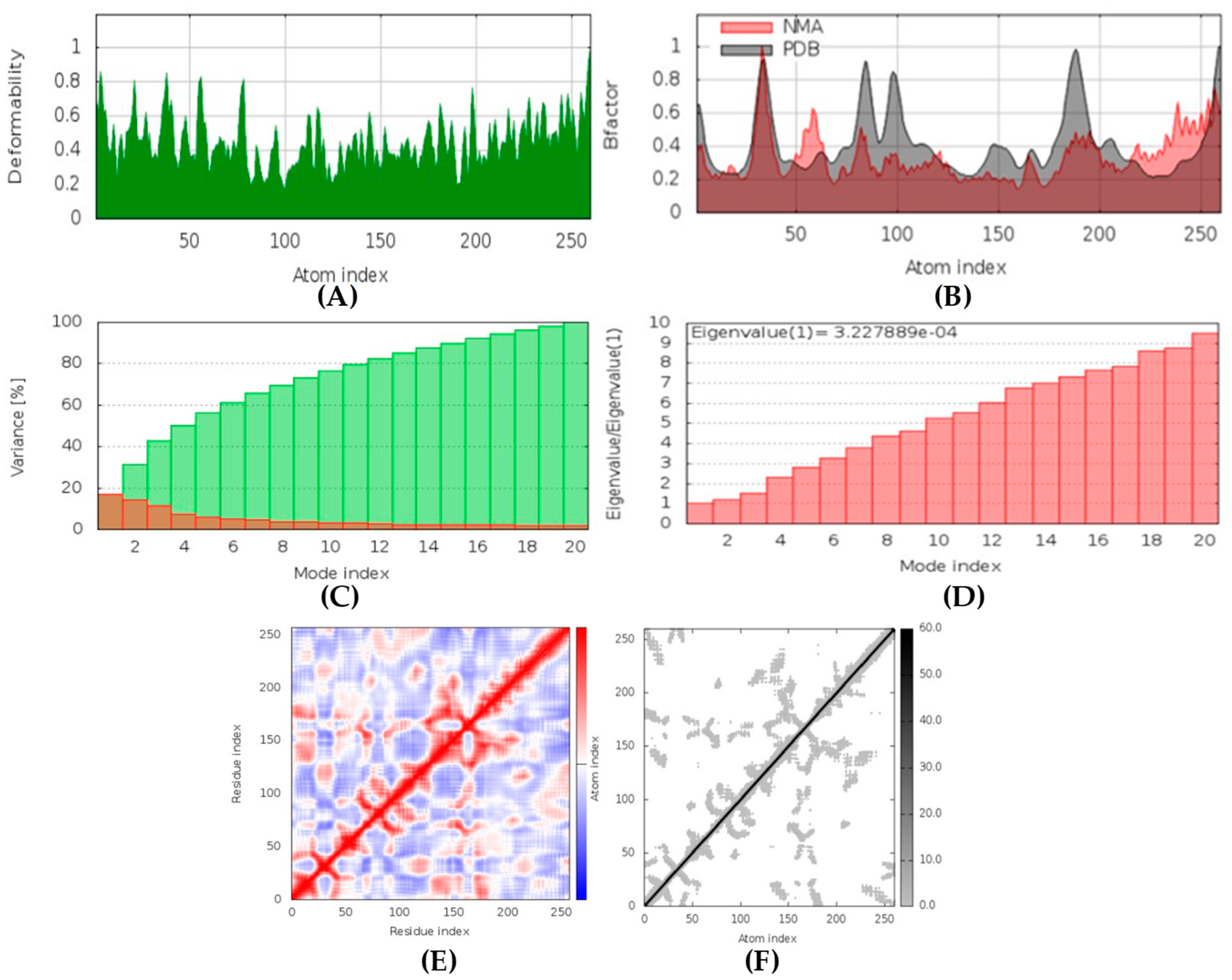
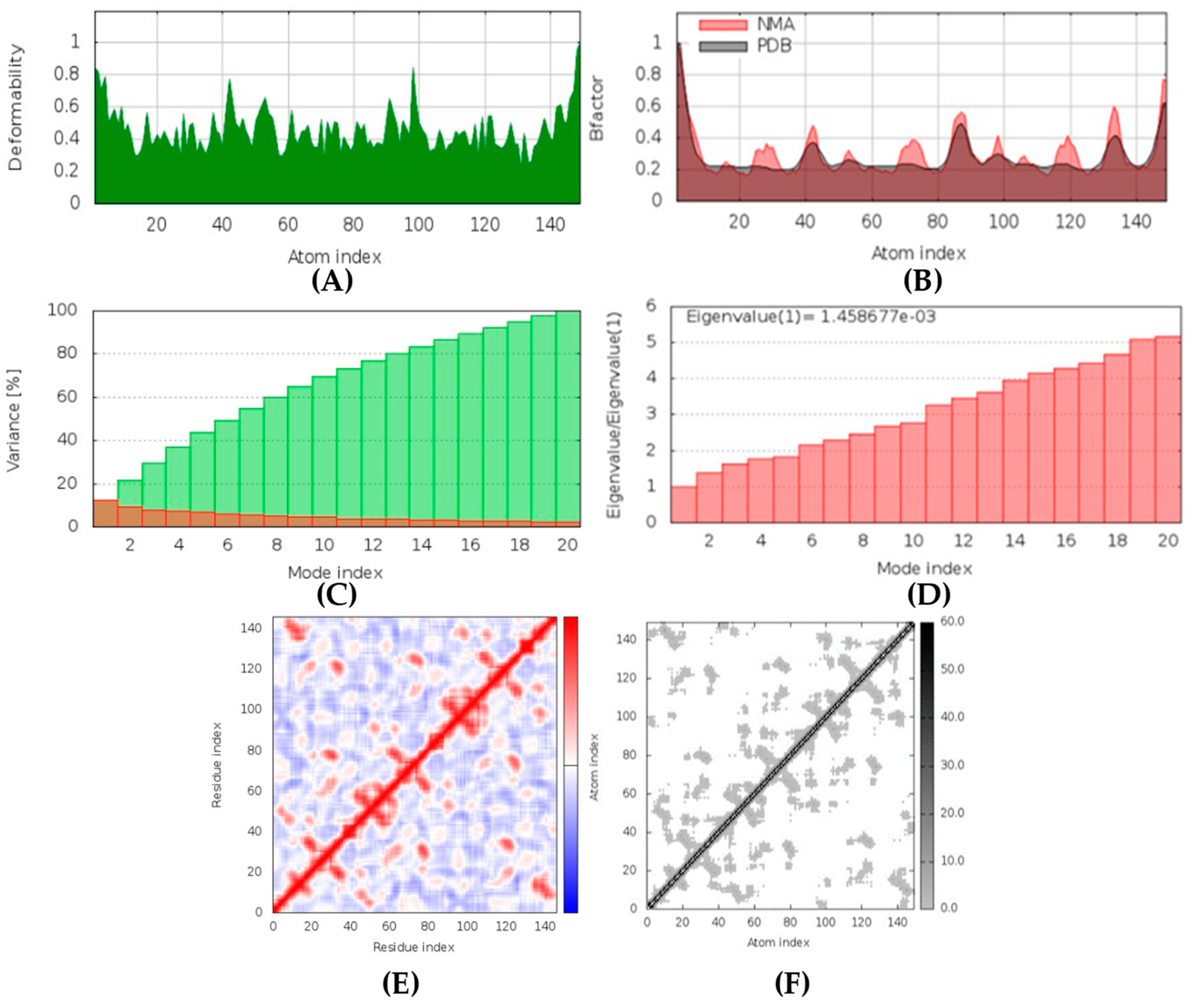
(+)-Curcudiol was found to be antifungal, anti-eczematic, antiviral, anti-infective, and antiseptic, and it is also a bronchodilator. In addition, it is histidine kinase, exo-1,3-beta-glucan synthase, RdRp, lipase, lanosterol 14 alpha-demethylase, and alpha and beta-glucuronidase inhibitor. This means it might inhibit six out of seven targets involved in this study. Molecular docking confirmed the suspicion; (+)-curcudiol had the highest binding affinity, −11.4 kcal/mol, with lanosterol 14 alpha-demethylase. MD simulation also suggested that the complex formed between the two was stable. Drug-likeness estimation suggested that (+)-curcudiol does not violate Lipinski rules: thus, it might have a drug-like tendency. Swiss-ADME indicated that it has moderate solubility and bioavailability, 0.55. High GI absorption indicated the ability to cross the blood-brain barrier. OSIRIS analysis showed (+)-curcudiol poses low to no risk of being an irritant, mutagenic agent, or tumorigenic agent, and it does not affect the reproductive system. PkCSM analysis suggests that (+)-curcudiol is non-hepatotoxic. ProTox-II indicates that it belongs to class 5. StopTox found that (+)-curcudiol might not cause acute inhalation or dermal and oral toxicity. However, it can cause skin sensitization. It is corrosive to the eyes and not the skin.
(+)-Curcuphenol was predicted to be antifungal, anti-eczematic, antiviral, anti-infective, anti-inflammatory, a bronchodilator, mucolytic, and antiseptic. In addition, (+)-curcuphenol is histidine kinase alpha and beta-glucuronidase, lanosterol 14 alpha-demethylase, lipase, exo-1,3-beta-glucan-synthase, RdRp, and rhizopuspepsin inhibitor. This compound has been found to have broad-spectrum activity, as it has the potential to inhibit all seven targets involved in this study. These properties could be very helpful in addressing the COVID pandemic era. Molecular docking studies showed that (+)-curcuphenol had a good binding affinity with fungal lipase target, −8 kcal/mol. However, MD simulation indicated that the complex formed might be slightly unstable. Drug-likeness estimation suggested that (+)-curcuphenol does not violate Lipinski rules, thus having drug-like tendencies. Swiss-ADME indicated that it has moderate solubility and bioavailability of 0.55. High GI absorption was indicated, with the ability to cross the blood-brain barrier. OSIRIS analysis showed (+)-curcuphenol can be an irritant. PkCSM analysis suggests that (+)-curcuphenol is non-hepatotoxic. ProTox-II indicates that it belongs to class 4. StopTox found that (+)-curcuphenol should not cause acute inhalation or dermal and oral toxicity, but it can cause skin sensitization. It can be corrosive to the eyes but not to the skin.
Tetillapyrone and nortetillapyrone are potentially antifungal, anti-eczematic, antiviral, and anti-infective. In addition, they are histidine kinase, beta-glucuronidase, RdRp, and exo-1,3-beta-glucan-synthase inhibitors. This means they can inhibit CotH3, mucoricin, and two other target proteins. They showed mostly average and some good interactions in docking studies in comparison to other compounds. Both compounds do not violate Lipinski’s rules, thus having drug-like tendencies. They have high solubility and bioavailability of 0.55. Although a high GI absorption was indicated, they cannot cross the blood-brain barrier. OSIRIS analysis showed they are both harmless. PkCSM analysis suggests that they both are non-hepatotoxic. ProTox-II indicates that they belong to class 3. StopTox found that they might not cause acute inhalation or dermal and oral toxicity. In addition, they may not cause skin sensitization and corrosion of skin and eyes.
Aurantosides I and K are predicted to be antifungal, antiviral, antibiotic, and anti-inflammatory. In addition, they can inhibit beta-glucuronidase and exo-1,3-beta-glucan-synthase enzymes. Docking results have shown that the interaction between aurantoside I and exo-1,3-beta-glucan-synthase obtained the highest binding affinity value, −11.4 kcal/mol. MD simulations have also indicated the formation of a stable complex. According to Swiss-ADME, they both have high solubility, and the bioavailability was lowest compared to all other compounds. As a result, the GI tract absorption is also found below. They are not blood-brain-barrier permeable. OSIRIS output suggests they are potential irritants and might affect the reproductive system. PkCSM indicates aurantoside I might be hepatotoxic. They belong to class 5 compounds in ProTox-II. StopTox found that they should not cause acute inhalation, dermal and oral toxicity, or skin sensitization. However, they can cause irritation and corrosion of the eyes.
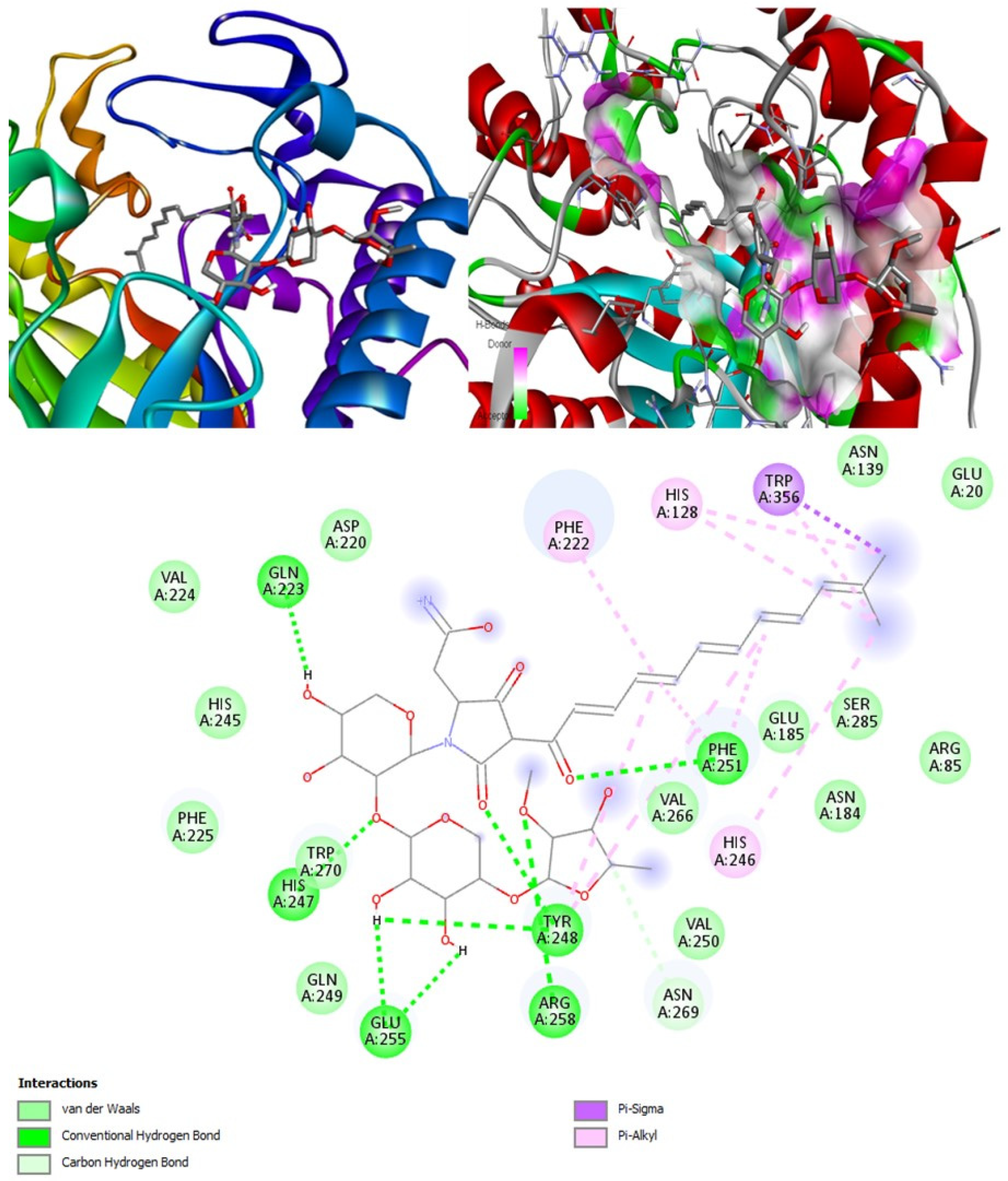
Concerning medical drugs in this study, amphotericin B was predicted to be an antifungal, antiviral, anti-inflammatory, and anti-infective enhancer. By contrast, isavuconazole and posaconazole are only antifungals. The mechanism of action of these drugs is destroying the fungal cell wall by inhibiting the synthesis of beta-glucan. Our calculations revealed that both isavuconazole and posaconazole inhibit a common target, lanosterol 14 alpha-demethylase, thus preventing the conversion of lanosterol into ergosterol, leading to the damage of the plasma membrane of the fungi. Additionally, isavuconazole might inhibit CotH3, thus being a kinase inhibitor, while posaconazole, in OSIRIS analysis, was found to be potentially mutagenic and tumorigenic.
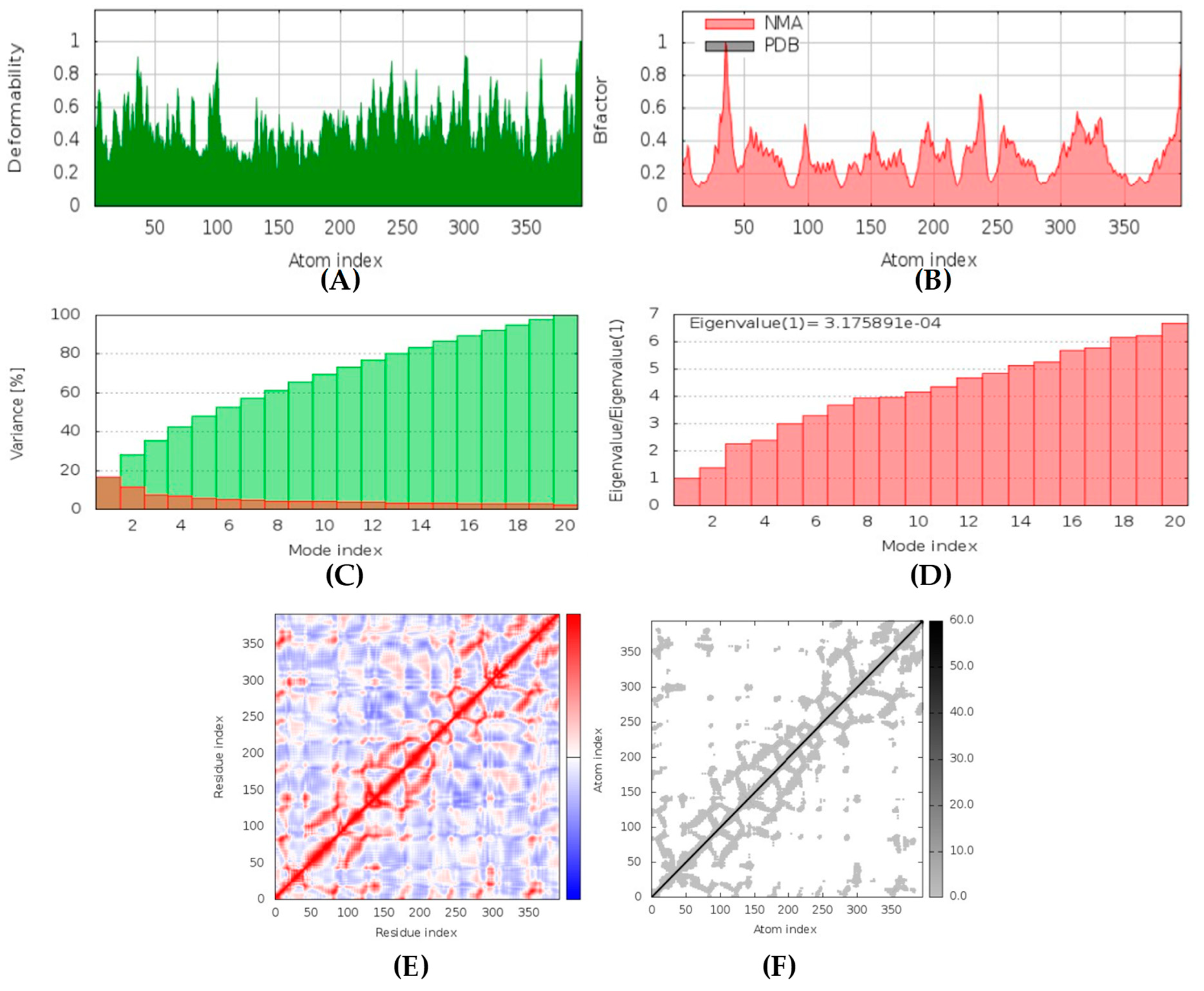
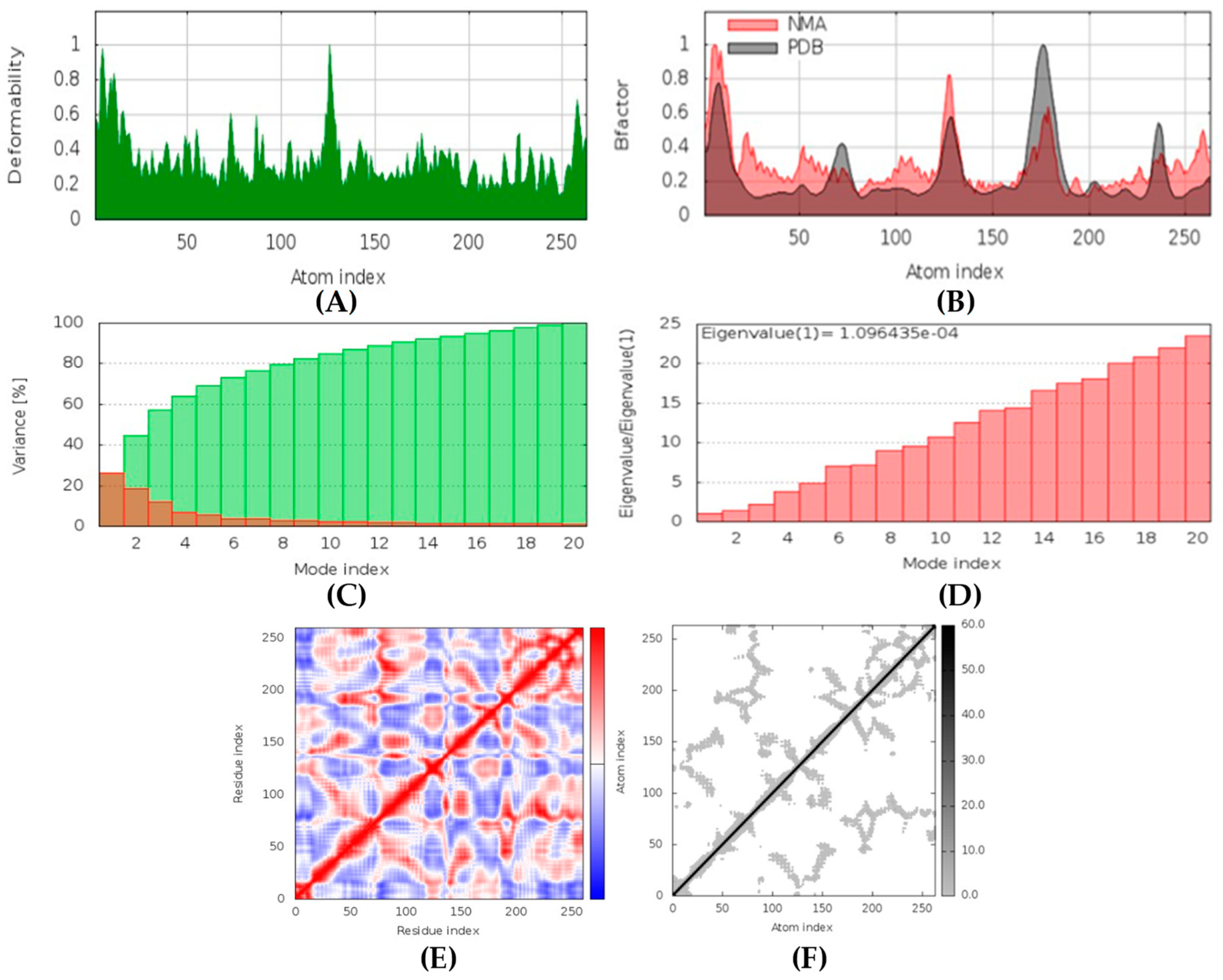
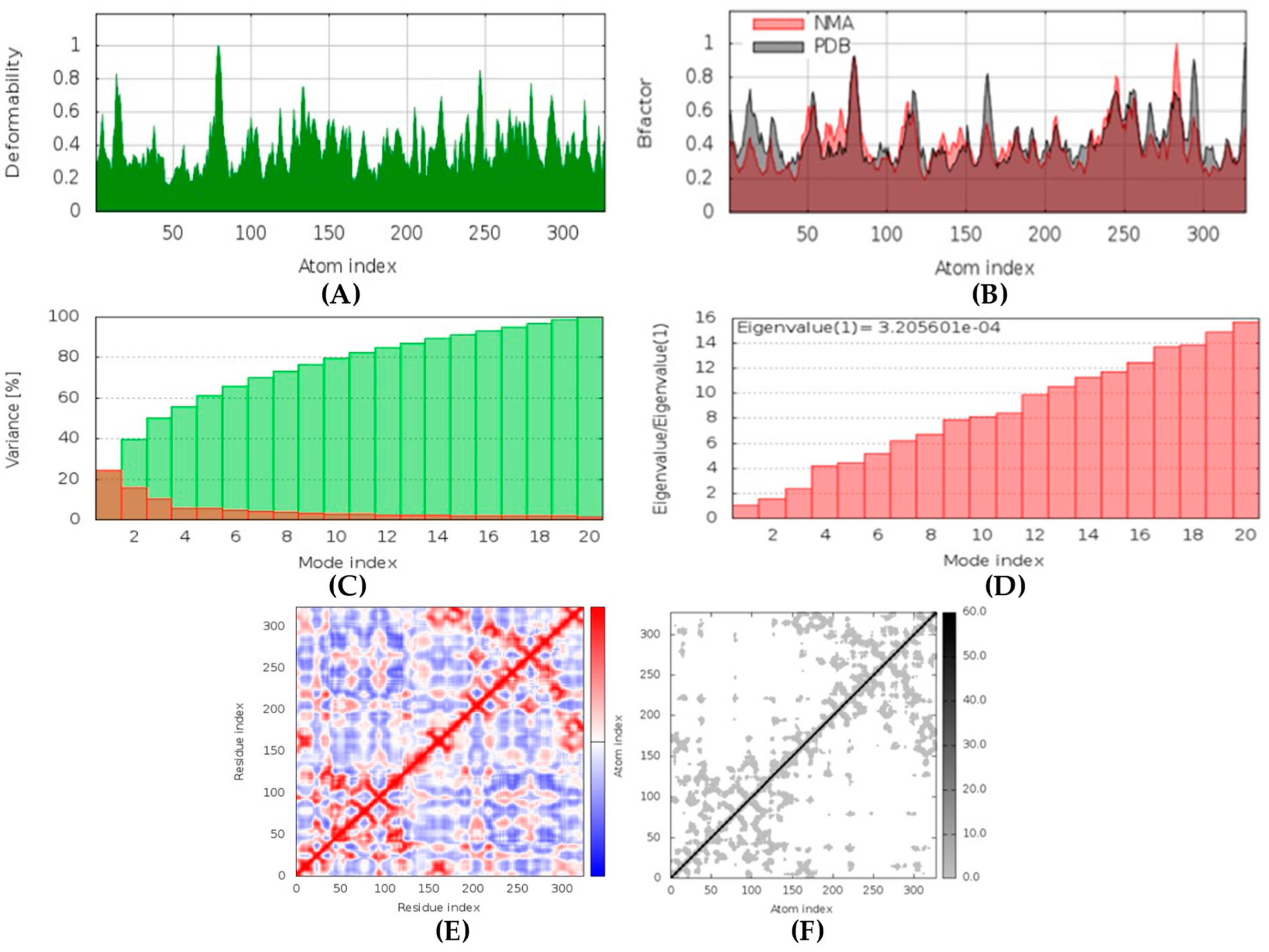
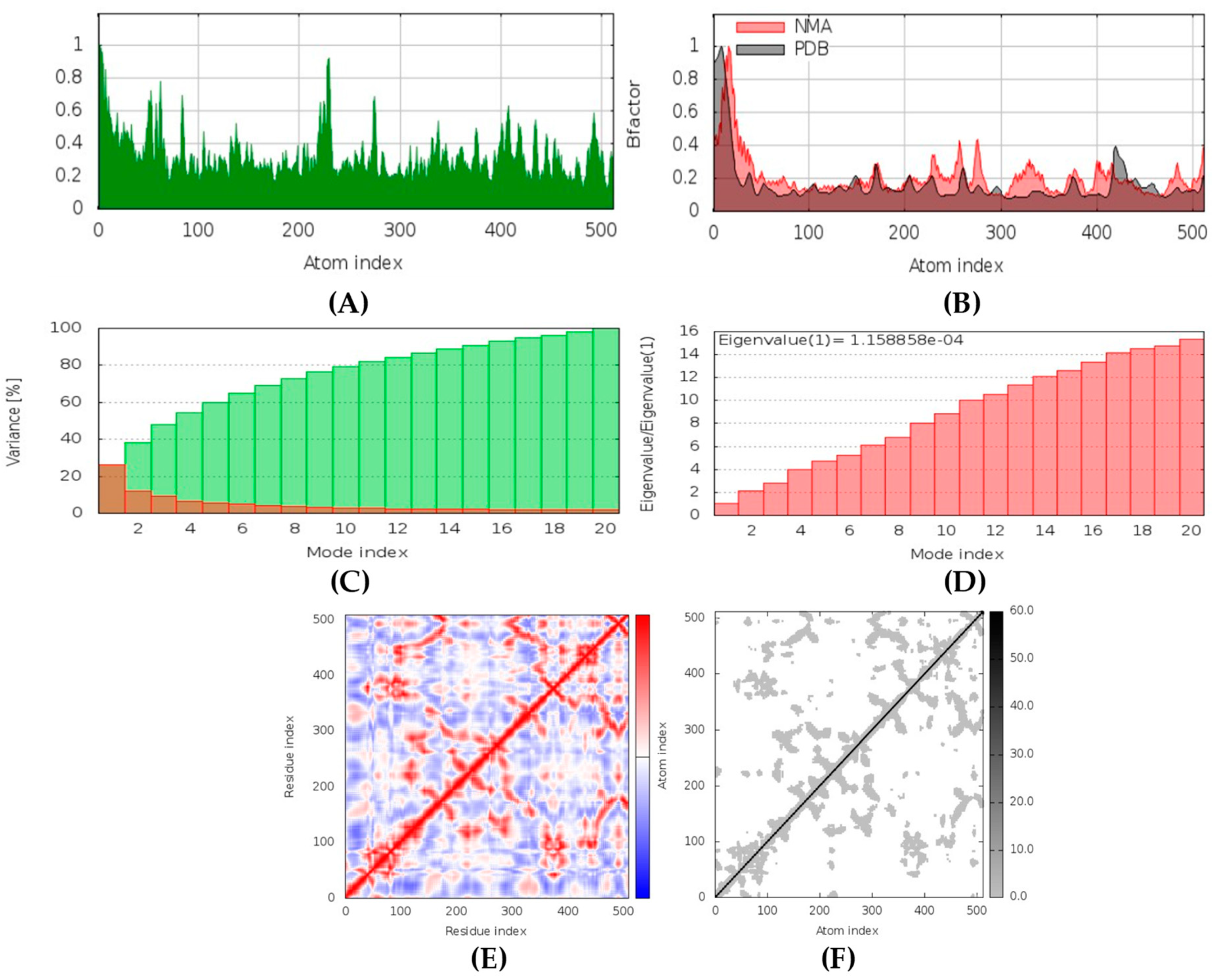
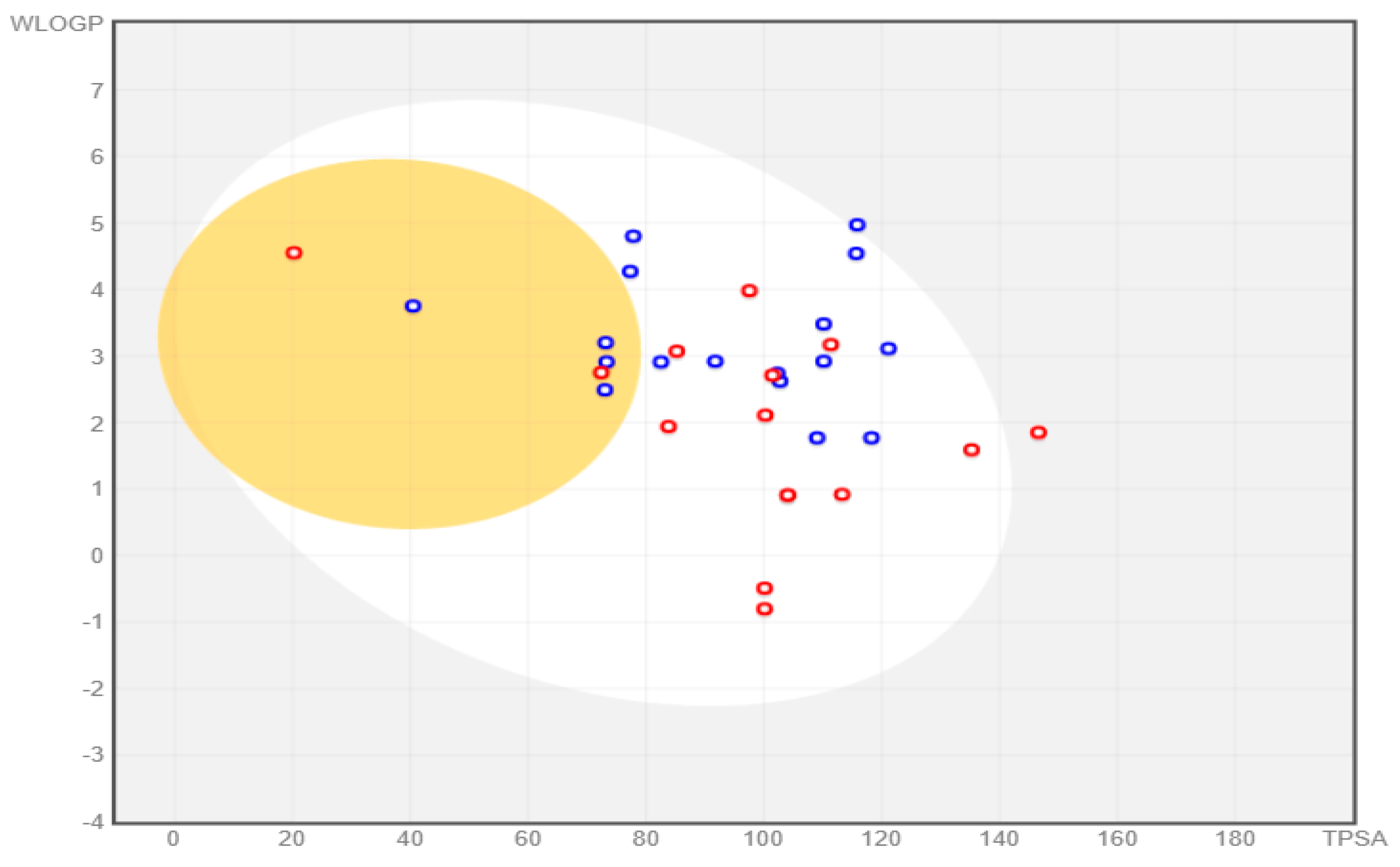
All the compounds were categorized based on target-specific biological activity (see
Table 4). The compounds in this study were shown to be capable of inhibiting different targets to varying degrees. We have found that some MNPs from sponges presented better target-specific inhibition as compared to the three antifungal drugs, highlighting their pharmaceutical potential. All 32 MNPs are worth investigating. However, (+)-curcudiol and (+)-curcuphenol showed promising potential to inhibit the majority of the fungal targets mentioned in this study. In addition, these MNPs showed satisfactory drug-likeness properties with the capability to permeate the blood-brain barrier. This is important as COVID-19-associated mucormycosis also affects the brain in some cases [
62]. Moreover, the findings of this study suggest that they might pose overall low toxicity risks. We recommend further laboratory interventions and experimental investigations to recognize the true capability of target inhibition by in vitro and in vivo studies.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





















