
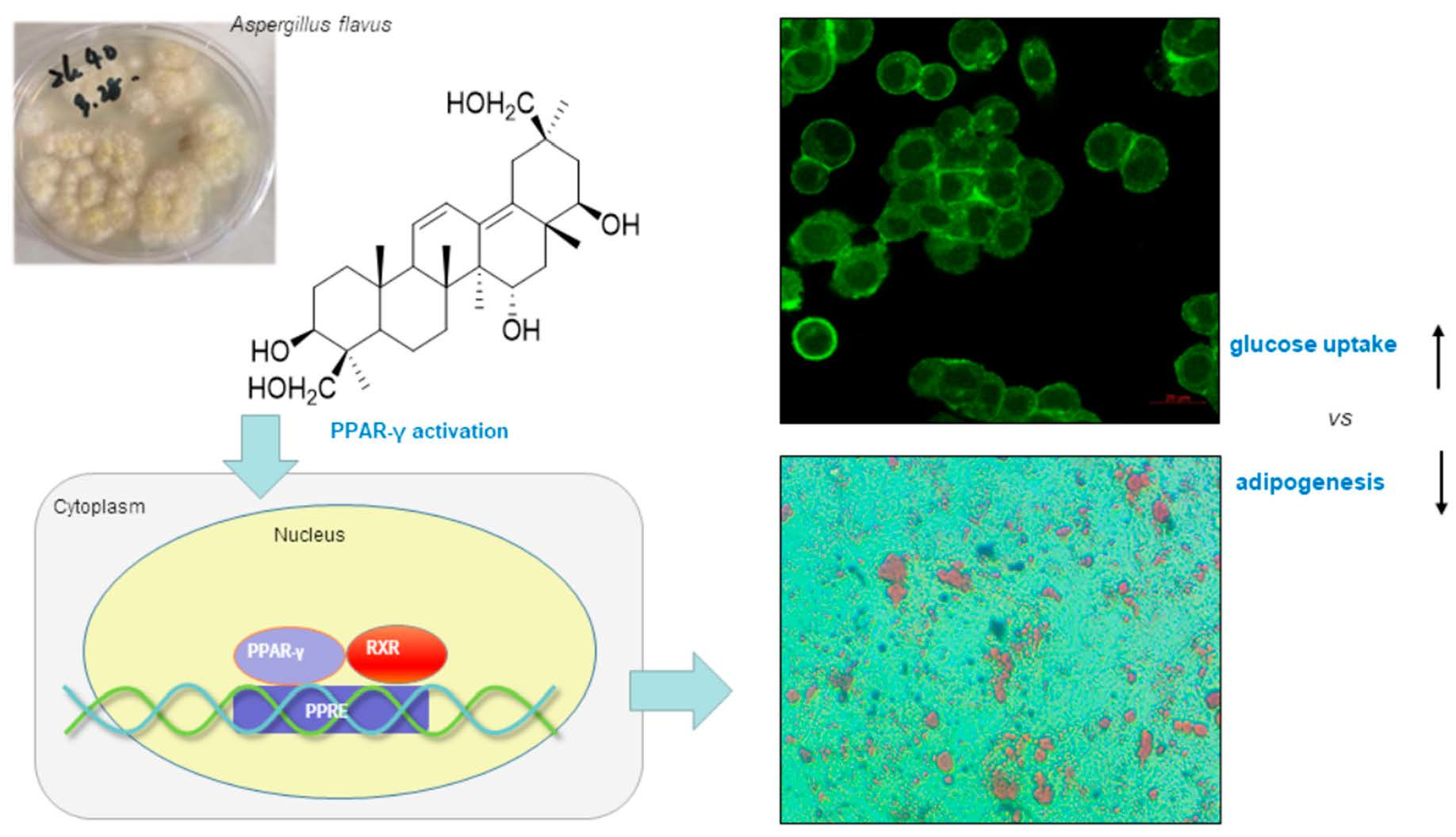
A New Fungal Triterpene from the Fungus Aspergillus flavus Stimulates Glucose Uptake without Fat Accumulation


Abstract
:
1. Introduction
2. Results and Discussion
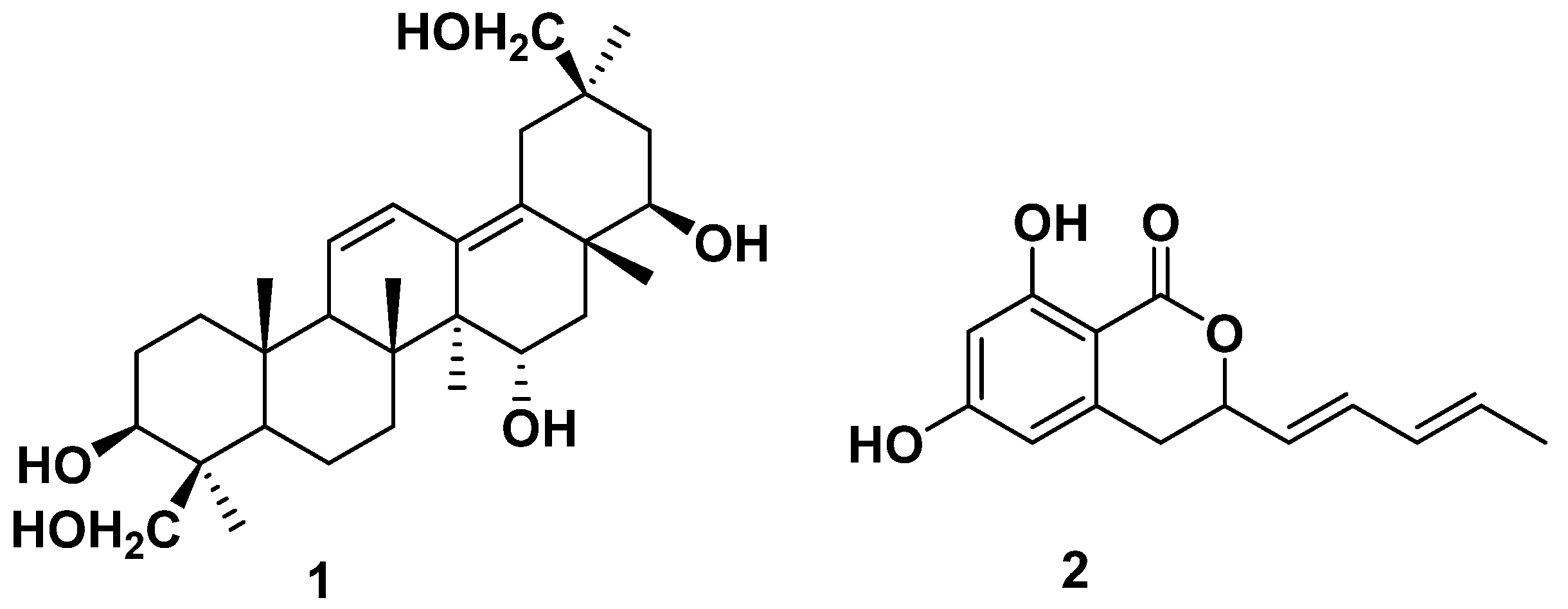
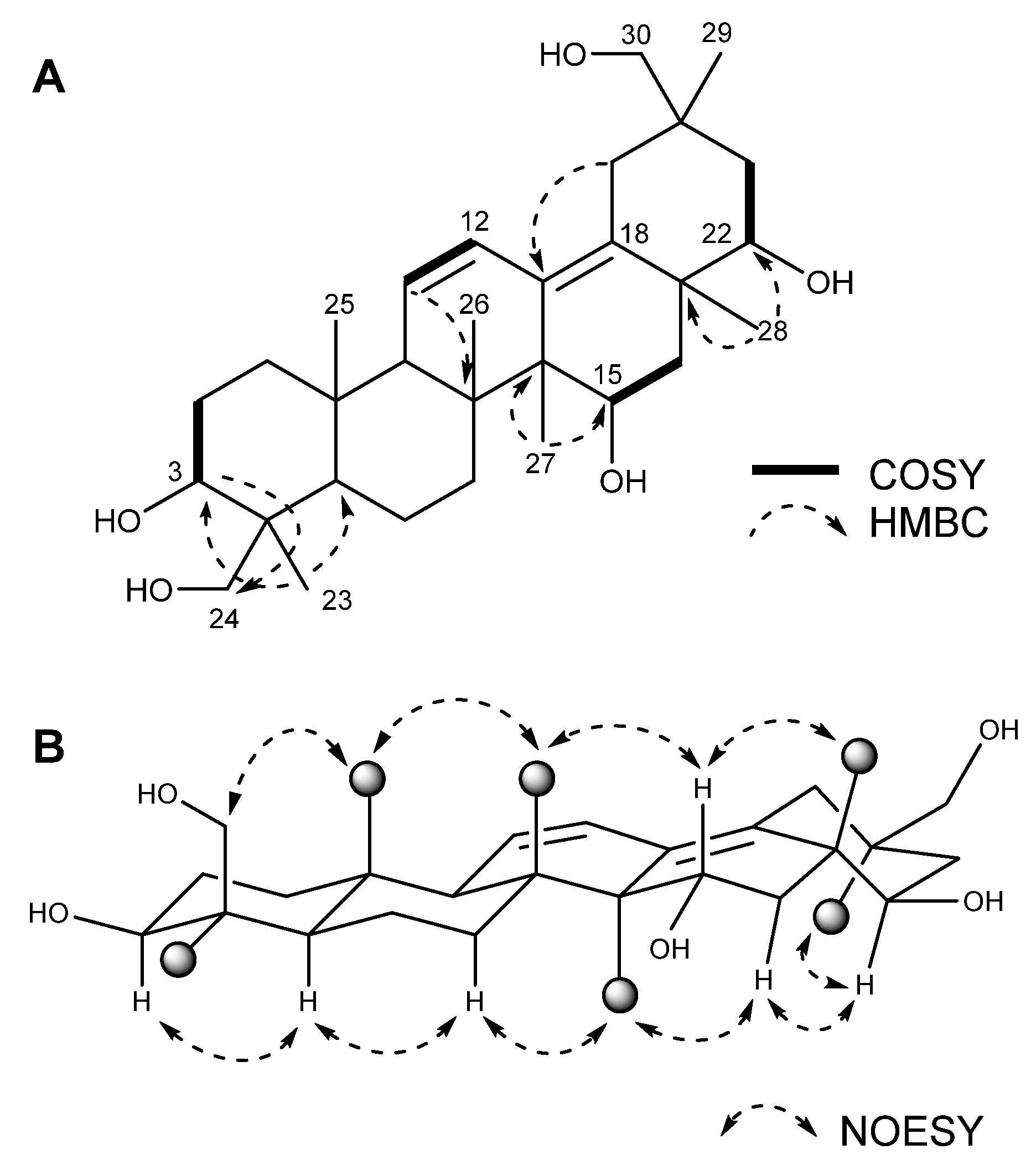
2.1. Structure of the New Compound
2.2. Identification of the Known Compounds
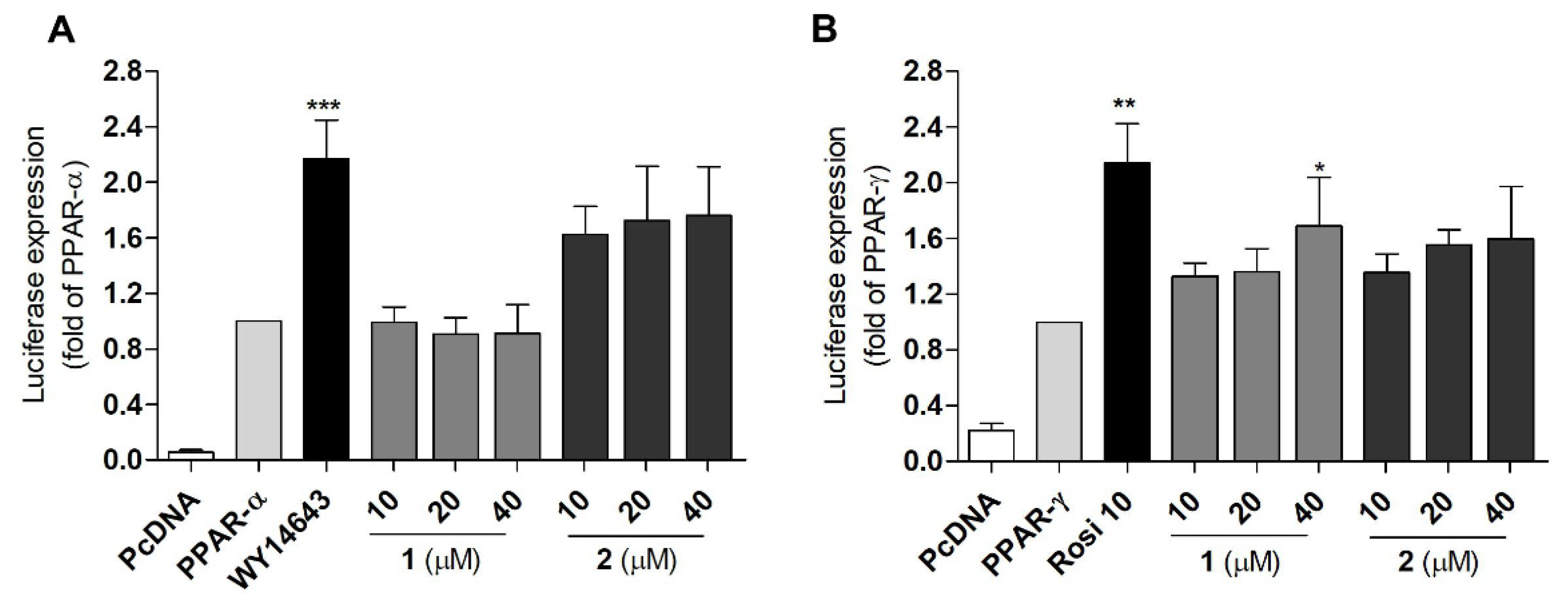
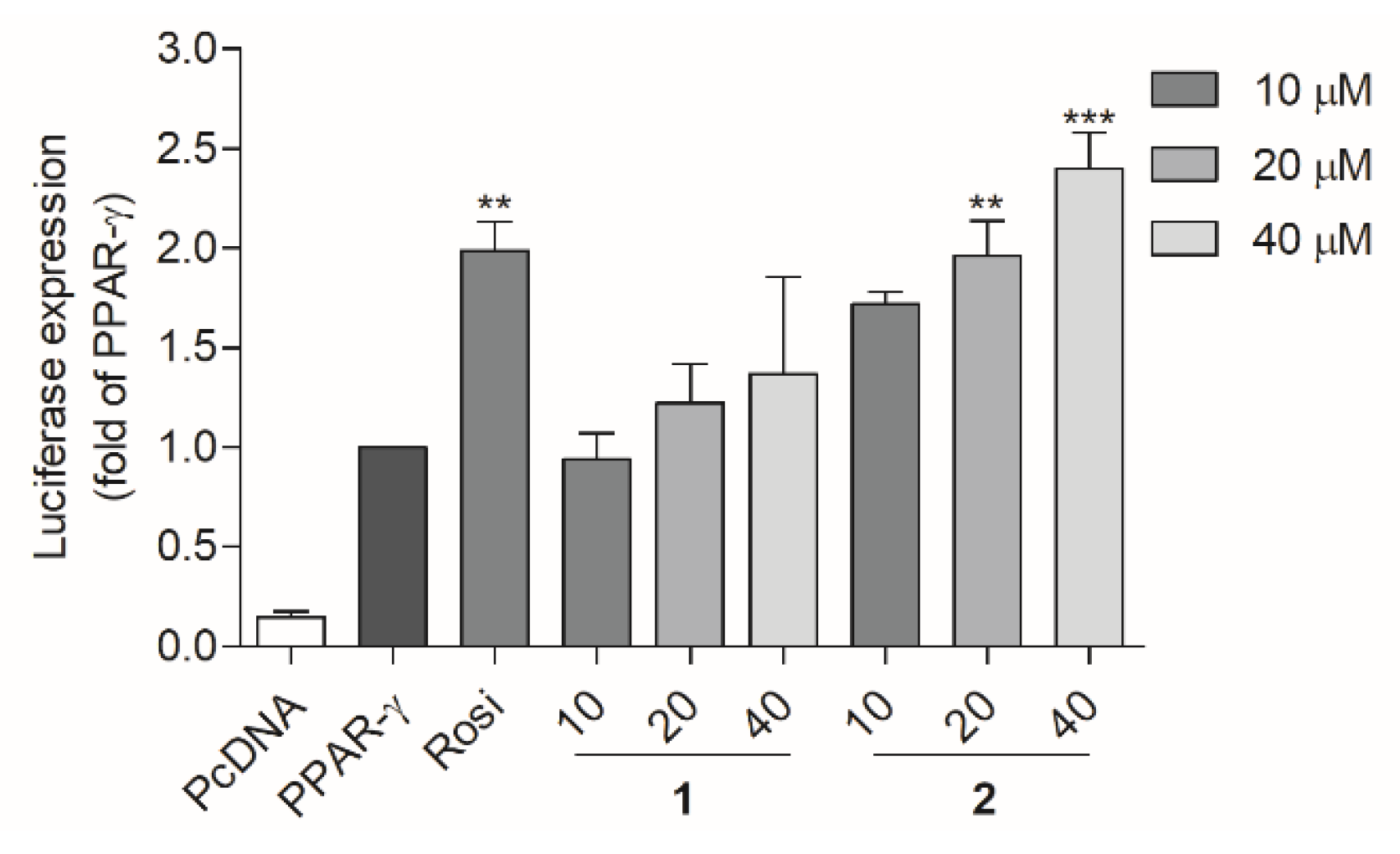
2.3. PPAR Transactivation by Compound 1 and 2
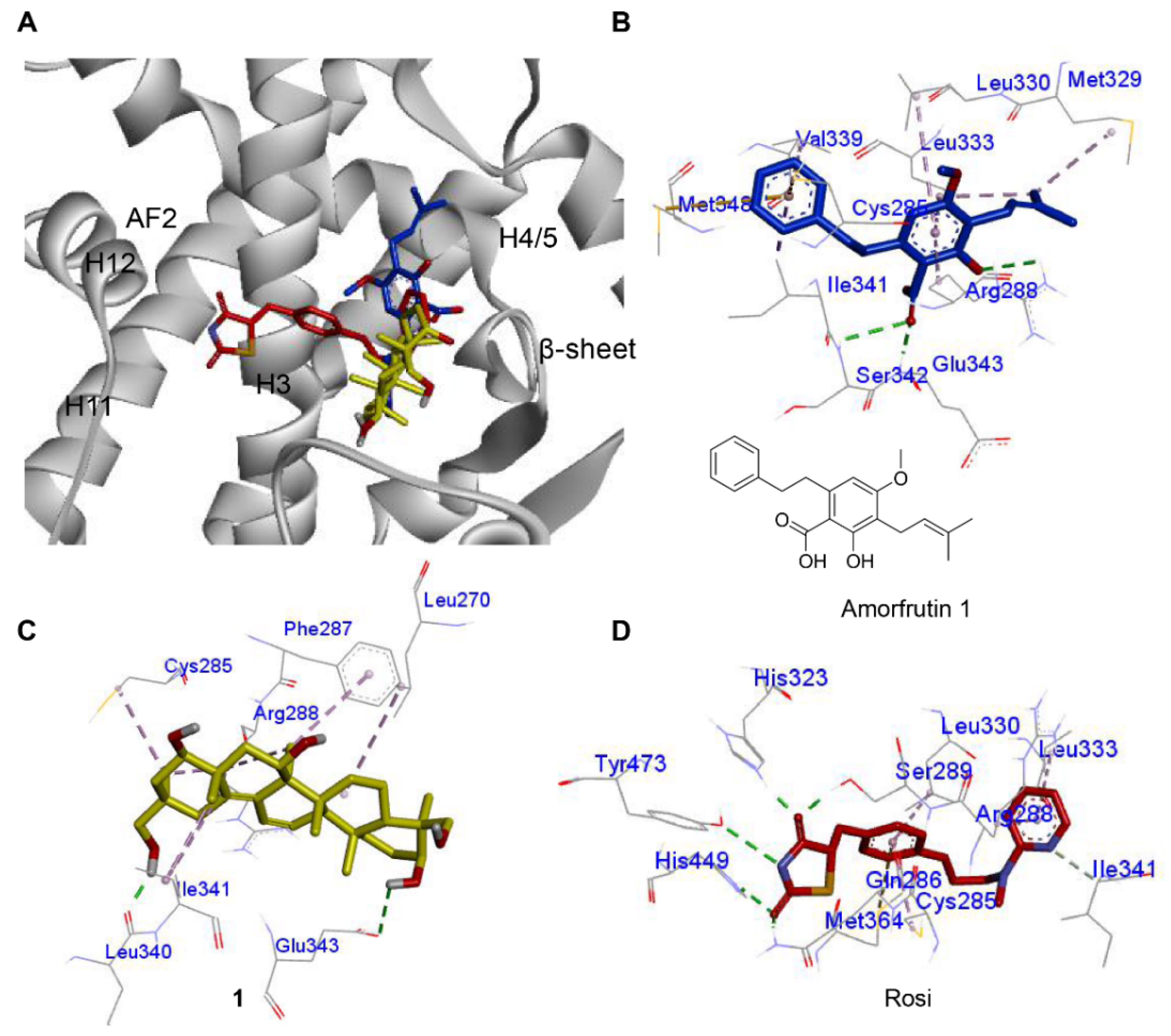
2.4. Molecular Docking of Compound 1 with PPAR-γ
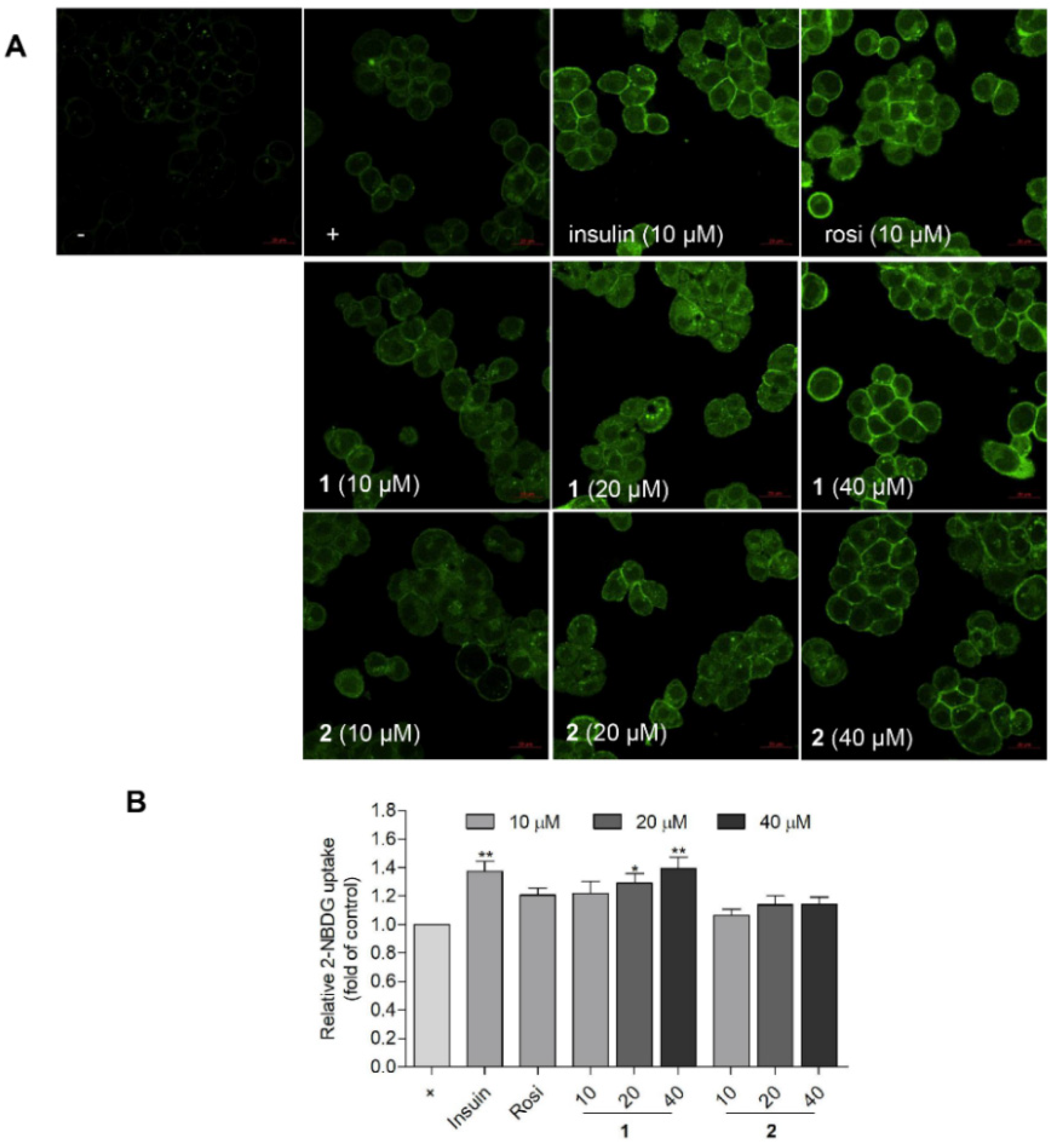
2.5. Compound 1 Stimulated the Glucose Uptake in HepG2 Human Liver Cells
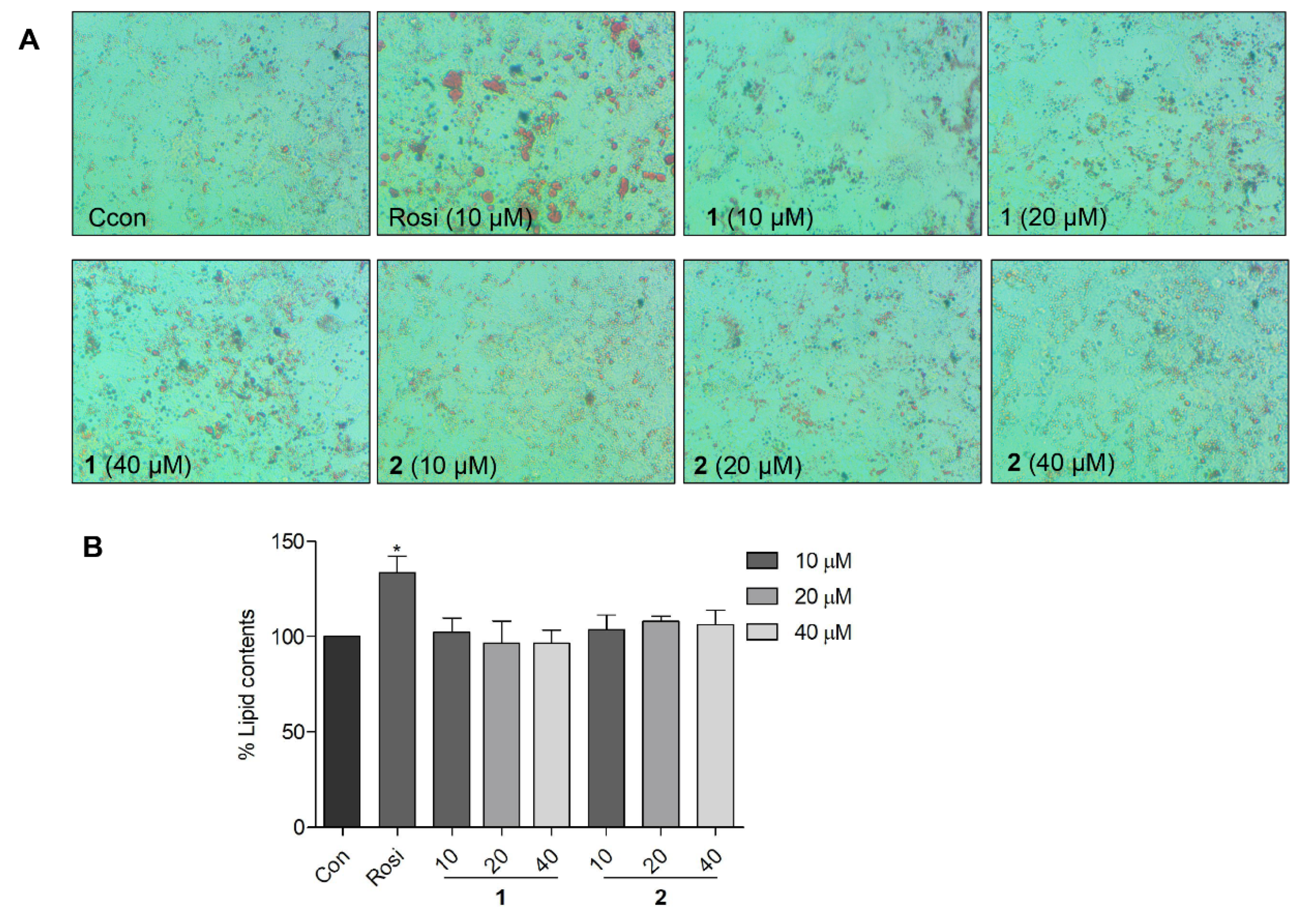
2.6. Effects of Compound 1 on Lipid Accumulation in 3T3-L1 Preadipocytes
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Materials
3.3. Extraction and Isolation
3.4. Computational Methods
3.5. Cell Culture and Cell Viability Assay
3.6. Luciferase Assay
3.7. Adipocyte Differentiation Assay
3.8. Glucose Uptake Assay
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Akter, R.; Afrose, A.; Rahman, M.R.; Chowdhury, R.; Nirzhor, S.S.R.; Khan, R.I.; Kabir, M.T. A comprehensive analysis into the therapeutic application of natural products as SIRT6 modulators in Alzheimer’s disease, aging, cancer, inflammation, and diabetes. Int. J. Mol. Sci. 2021, 22, 4180. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.-R. Marine natural products. Nat. Prod. Rep. 2021, 38, 362–413. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Romano, G.; Costantini, M.; Sansone, C.; Lauritano, C.; Ruocco, N.; Ianora, A. Marine microorganisms as a promising and sustainable source of bioactive molecules. Mar. Environ. Res. 2017, 128, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Gothai, S.; Ganesan, P.; Park, S.Y.; Fakurazi, S.; Choi, D.K.; Arulselvan, P. Natural phyto-bioactive compounds for the treatment of type 2 diabetes: Inflammation as a target. Nutrients 2016, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Jay, M.A.; Ren, J. Peroxisome proliferator-activated receptor (PPAR) in metabolic syndrome and type 2 diabetes mellitus. Curr. Diabetes Rev. 2007, 3, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Calkin, A.C.; Thomas, M.C. PPAR agonists and cardiovascular disease in diabetes. PPAR Res. 2008, 2008, 245410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Kim, H.i.; Ahn, Y.h. Role of peroxisome proliferator-activated receptor-γ in the glucose-sensing apparatus of liver and β-cells. Diabetes 2004, 53, S60–S65. [Google Scholar] [CrossRef] [Green Version]
- Wright, M.B.; Bortolini, M.; Tadayyon, M.; Bopst, M. Minireview: Challenges and opportunities in development of PPAR agonists. Mol. Endocrinol. 2014, 28, 1756–1768. [Google Scholar] [CrossRef] [Green Version]
- Blazer-Yost, B.L. PPARγ agonists: Blood pressure and edema. PPAR Res. 2010, 2010, 785369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgermeister, E.; Schnoebelen, A.; Flament, A.; Benz, J.R.; Stihle, M.; Gsell, B.; Rufer, A.; Ruf, A.; Kuhn, B.; Märki, H.P. A novel partial agonist of peroxisome proliferator-activated receptor-γ (PPARγ) recruits PPARγ-coactivator-1α, prevents triglyceride accumulation, and potentiates insulin signaling in vitro. Mol. Endocrinol. 2006, 20, 809–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motani, A.; Wang, Z.; Weiszmann, J.; McGee, L.R.; Lee, G.; Liu, Q.; Staunton, J.; Fang, Z.; Fuentes, H.; Lindstrom, M. INT131: A selective modulator of PPARγ. J. Mol. Biol. 2009, 386, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Bruning, J.B.; Chalmers, M.J.; Prasad, S.; Busby, S.A.; Kamenecka, T.M.; He, Y.; Nettles, K.W.; Griffin, P.R. Partial agonists activate PPARγ using a helix 12 independent mechanism. Structure 2007, 15, 1258–1271. [Google Scholar] [CrossRef] [PubMed]
- Montanari, R.; Saccoccia, F.; Scotti, E.; Crestani, M.; Godio, C.; Gilardi, F.; Loiodice, F.; Fracchiolla, G.; Laghezza, A.; Tortorella, P. Crystal structure of the peroxisome proliferator-activated receptor γ (PPARγ) ligand binding domain complexed with a novel partial agonist: A new region of the hydrophobic pocket could be exploited for drug design. J. Med. Chem. 2008, 51, 7768–7776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Lavan, B.E.; Gregoire, F.M. Selective modulators of PPAR-γ activity: Molecular aspects related to obesity and side-effects. PPAR Res. 2007, 2007, 32696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidner, C.; de Groot, J.C.; Prasad, A.; Freiwald, A.; Quedenau, C.; Kliem, M.; Witzke, A.; Kodelja, V.; Han, C.T.; Giegold, S.; et al. Amorfrutins are potent antidiabetic dietary natural products. Proc. Natl. Acad. Sci. USA 2012, 109, 7257–7262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Vallve, S.; Guasch, L.; Tomas-Hernandez, S.; del Bas, J.M.; Ollendorff, V.; Arola, L.; Pujadas, G.; Mulero, M. Peroxisome proliferator-activated receptor γ (PPARγ) and ligand choreography: Newcomers take the stage: Miniperspective. J. Med. Chem. 2015, 58, 5381–5394. [Google Scholar] [CrossRef]
- Li, D.d.; Wang, Y.; Kim, E.L.; Hong, J.; Jung, J.H. Neuroprotective effect of cyclo-(L-Pro-L-Phe) isolated from the jellyfish-derived fungus Aspergillus flavus. Mar. Drugs 2021, 19, 417. [Google Scholar] [CrossRef]
- Lan, W.J.; Fu, S.J.; Xu, M.Y.; Liang, W.L.; Lam, C.K.; Zhong, G.H.; Xu, J.; Yang, D.P.; Li, H.J. Five new cytotoxic metabolites from the marine fungus neosartorya pseudofischeri. Mar. Drugs 2016, 14, 18. [Google Scholar] [CrossRef] [Green Version]
- Elnaggar, M.S.; Ebada, S.S.; Ashour, M.L.; Ebrahim, W.; Singab, A.; Lin, W.; Liu, Z.; Proksch, P. Two new triterpenoids and a new naphthoquinone derivative isolated from a hard coral-derived fungus Scopulariopsis sp. Fitoterapia 2017, 116, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Asada, M.; Amagaya, S.; Takai, M.; Ogihara, Y. New triterpenoids from the leaves of Tetrapanax papyriferum. J. Chem. Soc. 1980, 325–329. [Google Scholar] [CrossRef]
- Lee, D.S.; Eom, S.H.; Jeong, S.Y.; Shin, H.J.; Je, J.Y.; Lee, E.W.; Chung, Y.H.; Kim, Y.M.; Kang, C.K.; Lee, M.S. Anti-methicillin-resistant Staphylococcus aureus (MRSA) substance from the marine bacterium pseudomonas sp. UJ-6. Environ. Toxicol. Pharmacol. 2013, 35, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Y.; Sattler, I.; Deng, Z.W.; Groth, I.; Walther, G.; Menzel, K.D.; Peschel, G.; Grabley, S.; Lin, W.H. A-seco-oleane-type triterpenes from Phomopsis sp (strain HK10458) isolated from the mangrove plant Hibiscus tiliaceus. Phytochemistry 2008, 69, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Smetanina, O.F.; Kuznetzova, T.A.; Denisenko, V.A.; Pivkin, M.V.; Khudyakova, Y.V.; Gerasimenko, A.V.; Popov, D.Y.; Il’in, S.G.; Elyakov, G.B. 3β-Methoxyolean-18-ene (miliacin) from the marine fungus Chaetomium olivaceum. Russ. Chem. Bull. 2001, 50, 2463–2465. [Google Scholar] [CrossRef]
- Zhang, H.; Saurav, K.; Yu, Z.; Mandi, A.; Kurtan, T.; Li, J.; Tian, X.; Zhang, Q.; Zhang, W.; Zhang, C. α-Pyrones with diverse hydroxy substitutions from three marine-derived nocardiopsis strains. J. Nat. Prod. 2016, 79, 1610–1618. [Google Scholar] [CrossRef] [PubMed]
- Li, S.R.; Chen, P.Y.; Chen, L.Y.; Lo, Y.F.; Tsai, I.L.; Wang, E.C. Synthesis of haginin E, equol, daidzein, and formononetin from resorcinol via an isoflavene intermediate. Tetrahedron Lett. 2009, 50, 2121–2123. [Google Scholar] [CrossRef]
- Boryski, J.; Grynkiewicz, G. A regioselective synthesis of genistein 4’-O-ribofuranosides. Synthesis 2001, 2001, 2170–2174. [Google Scholar] [CrossRef]
- Pouysegu, L.; Marguerit, M.; Gagnepain, J.; Lyvinec, G.; Eatherton, A.J.; Quideau, S. Total synthesis of wasabidienones B1 and B0 via SIBX-mediated hydroxylative phenol dearomatization. Org. Lett. 2008, 10, 5211–5214. [Google Scholar] [CrossRef]
- Yoshimoto, Y.; Sawa, T.; Naganawa, H.; Sugai, T.; Takeuchi, T.; Imoto, M. MK800-62F1, a new inhibitor of apoptotic cell death, from streptomyces diastatochromogenes MK800-62F1 II. structure elucidation. J. Antibiot. 2000, 53, 575–578. [Google Scholar] [CrossRef] [Green Version]
- Mbafor, J.T.; Ndom, J.C.; Fomum, Z.T. Triterpenoid saponins from erythrina sigmoidea. Phytochemistry 1997, 44, 1151–1155. [Google Scholar] [CrossRef]
- Kitagawa, I.; Saito, M.; Taniyama, T.; Yoshikawa, M. Saponin and sapogenol. XXXVIII. Structure of soyasaponin A2, a bisdesmoside of soyasapogenol A, from soybean, the seeds of glycine max merrill. Chem. Pharm. Bull. 1985, 33, 598–608. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, I.; Furlong, K.; Flood, J.; Treat, V.P.; Goldstein, B.J. Dual PPAR α/γ agonists: Promises and pitfalls in type 2 diabetes. Am. J. Ther. 2007, 14, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Rigano, D.; Sirignano, C.; Taglialatela-Scafati, O. The potential of natural products for targeting PPARα. Acta Pharm. Sin. B 2017, 7, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Medjakovic, S.; Mueller, M.; Jungbauer, A. Potential health-modulating effects of isoflavones and metabolites via activation of PPAR and AhR. Nutrients 2010, 2, 241–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanova, M.; You, L.; Gaido, K.W.; Archibeque-Engle, S.; Janszen, D.B.; Heck, H.d.A. Developmental effects of dietary phytoestrogens in sprague-dawley rats and interactions of genistein and daidzein with rat estrogen receptors alpha and beta in vitro. Toxicol. Sci. 1999, 51, 236–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, Z. Dose-dependent effects of soy phyto-oestrogen genistein on adipocytes: Mechanisms of action. Obes. Rev. 2009, 10, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-γ. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Rines, A.K.; Sharabi, K.; Tavares, C.D.; Puigserver, P. Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nat. Rev. Drug Discov. 2016, 15, 786–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48, e218. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Wang, D.; Zhao, W.; Xu, L. Deciphering the roles of PPARγ in adipocytes via dynamic change of transcription complex. Front. Endocrinol. 2018, 9, 473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montanari, R.; Capelli, D.; Tava, A.; Galli, A.; Laghezza, A.; Tortorella, P.; Loiodice, F.; Pochetti, G. Screening of saponins and sapogenins from medicago species as potential PPARγ agonists and X-ray structure of the complex PPARγ/caulophyllogenin. Sci. Rep. 2016, 6, 27658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.L.; Han, L.F.; Yu, H.S.; Sang, M.M.; Liu, E.W.; Zhang, Y.; Fang, S.M.; Wang, T.; Gao, X.M. Analysis of the constituents in “Zhu She Yong Xue Shuan Tong” by ultra high performance liquid chromatography with quadrupole time-of-flight mass spectrometry combined with preparative high performance liquid chromatography. Molecules 2015, 20, 20518–20537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eom, S.H.; Liu, S.; Su, M.; Noh, T.H.; Hong, J.; Kim, N.D.; Chung, H.Y.; Yang, M.H.; Jung, J.H. Synthesis of phthalimide derivatives as potential PPAR-γ ligands. Mar. Drugs 2016, 14, 112. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | δH, Mult (J in Hz) | δC | No | δH, Mult (J in Hz) | δC |
|---|---|---|---|---|---|
| 1 | 1.95 (overlapped) 1.66 (overlapped) | 39.1 | 16 | 1.91 (overlapped) 1.60 (overlapped) | 43.9 |
| 2 | 1.85 (overlapped) 1.76 (overlapped) | 28.3 | 17 | 43.6 | |
| 3 | 3.38, dd (14.0, 4.9) | 81.2 | 18 | 137.2 | |
| 4 | 43.6 | 19 | 2.36, d (14.7) 1.85, d (14.7) | 32.9 | |
| 5 | 0.95 (overlapped) | 56.6 | 20 | 38.2 | |
| 6 | 1.73, m 1.46, m | 20.1 | 21 | 1.62 (overlapped) 1.36 (overlapped) | 38.8 |
| 7 | 1.63, m | 36.9 | 22 | 3.44, dd (10.0, 5.0) | 77.5 |
| 8 | 42.7 | 23 | 1.22, s | 23.0 | |
| 9 | 1.94 (overlapped) | 55.6 | 24 | 4.09, d (11.0) 3.42, d (11.0) | 65.0 |
| 10 | 37.7 | 25 | 0.92, s | 18.8 | |
| 11 | 5.64, d (10.5) * | 127.6 | 26 | 0.77, s | 17.8 |
| 12 | 6.36, dd (10.5, 3.0) * | 127.4 | 27 | 1.00, s | 14.9 |
| 13 | 136.8 | 28 | 1.09, s | 19.1 | |
| 14 | 48.8 | 29 | 0.80, s | 20.9 | |
| 15 | 4.03, dd (12.5, 4.1) | 66.7 | 30 | 3.28, s | 73.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.-d.; Wang, Y.; Kim, E.L.; Hong, J.; Jung, J.H. A New Fungal Triterpene from the Fungus Aspergillus flavus Stimulates Glucose Uptake without Fat Accumulation. Mar. Drugs 2022, 20, 203. https://doi.org/10.3390/md20030203
Li D-d, Wang Y, Kim EL, Hong J, Jung JH. A New Fungal Triterpene from the Fungus Aspergillus flavus Stimulates Glucose Uptake without Fat Accumulation. Marine Drugs. 2022; 20(3):203. https://doi.org/10.3390/md20030203
Chicago/Turabian StyleLi, Dan-dan, Ying Wang, Eun La Kim, Jongki Hong, and Jee H. Jung. 2022. "A New Fungal Triterpene from the Fungus Aspergillus flavus Stimulates Glucose Uptake without Fat Accumulation" Marine Drugs 20, no. 3: 203. https://doi.org/10.3390/md20030203