3.1. Conceptual DFT-Based Computational Peptidology
The quality of the chosen density function may be realized by comparing its results with results from high-level calculations or from experiential values. Nevertheless, this comparison is not always computationally practicable because of the large size of the molecules or the lack of experimental results for the chemical methods being explored. Our research group has developed a methodology known as KID [
15,
16,
17,
18,
19] in order to evaluate a particular density functional with regard to its internal coherence. It is evident that within the Generalized Kohn-Sham (GKS) version of DFT, some relationships exist between the KID methodology and the ionization energy theorem, which is a corollary of the Janak theorem [
20,
21,
22,
23,
24]. This is done by connecting
to −I and
to −A, through
Another KID descriptor
SL related to the difference in energies between the SOMO and the LUMO of the neutral system has been devised to aid in the verification of the accuracy of the methodology. In this work, a new Global KID Descriptor has ben defined as
whose value must be zero for the exact density functional meaning that it verifies the Ionization Energy theorem.
The MN12SX density functional has been shown to have a Koopmans-compliant behavior in earlier peptides studies. However, for a further validation of these model chemistry in the prediction of the chemical reactivity properties of the Veraguamides A–G, additional research is necessary. The CDFT software tool was used to make this determination, and the findings are shown in
Table 1, where the GKD has been calculated for all the cyclic peptides using several density functionals and in the presence of solvents of diverse polarity. A recent study [
24] has contrasted such behavior with a group of density functionals that includes the usual B3LYP [
62,
63,
64] and PBE0 [
65,
66] density functionals, the local density functionals BLYP [
63,
64,
67,
68] and PBE [
69] together with their long-range corrected variants, LC-BLYP and LC-PBE [
70], three longe-range corrected density functionals, CAM-B3LYP [
71], LC-
HPBE [
72] and
B97XD [
73], as well as three recently proposed density functionals, RSX-PBE, RSX-PBE0 and RSX-PBE0-1/3 [
74]. In order to attain completeness,
Table 1 shows a comparison of the fulfillment of the ionization energy theorem between the aforementioned density functionals and the screened-exchange MN12SX density functional used in this and previous cyclopeptides research in terms of the dielectric constant
of different common solvents:
The results from
Table 1 are very interesting because they show that there are various degrees in the fulfillment of the Janak and ionization energy theorems for the different long-range corrected density functionals involved. Moreover, it can be seen that the agreement varies depending on the dielectric constant of the solvent. Thus, it is possible to see that the CAM-B3LYP and
B97XD density functionals will be the most accurate when the study of the chemical reactivity is performed in the gas phase, that is, within the absence of any solvent. On the contrary, the MN12SX density functional will be superb when the conceptual DFT chemical reactivity properties estimated in terms of the frontier orbital energies are evaluated in the presence of polar solvents, mainly water, methanol and ethanol. As the chemical and biological processes in which the peptides are involved when they act as therapeutic drugs take part in the presence of water, the results from
Table 1 clearly demonstrate the superiority of the MN12SX/Def2TZVP/H2O model chemistry for the study of the molecules like the ones that are being considered through this research.
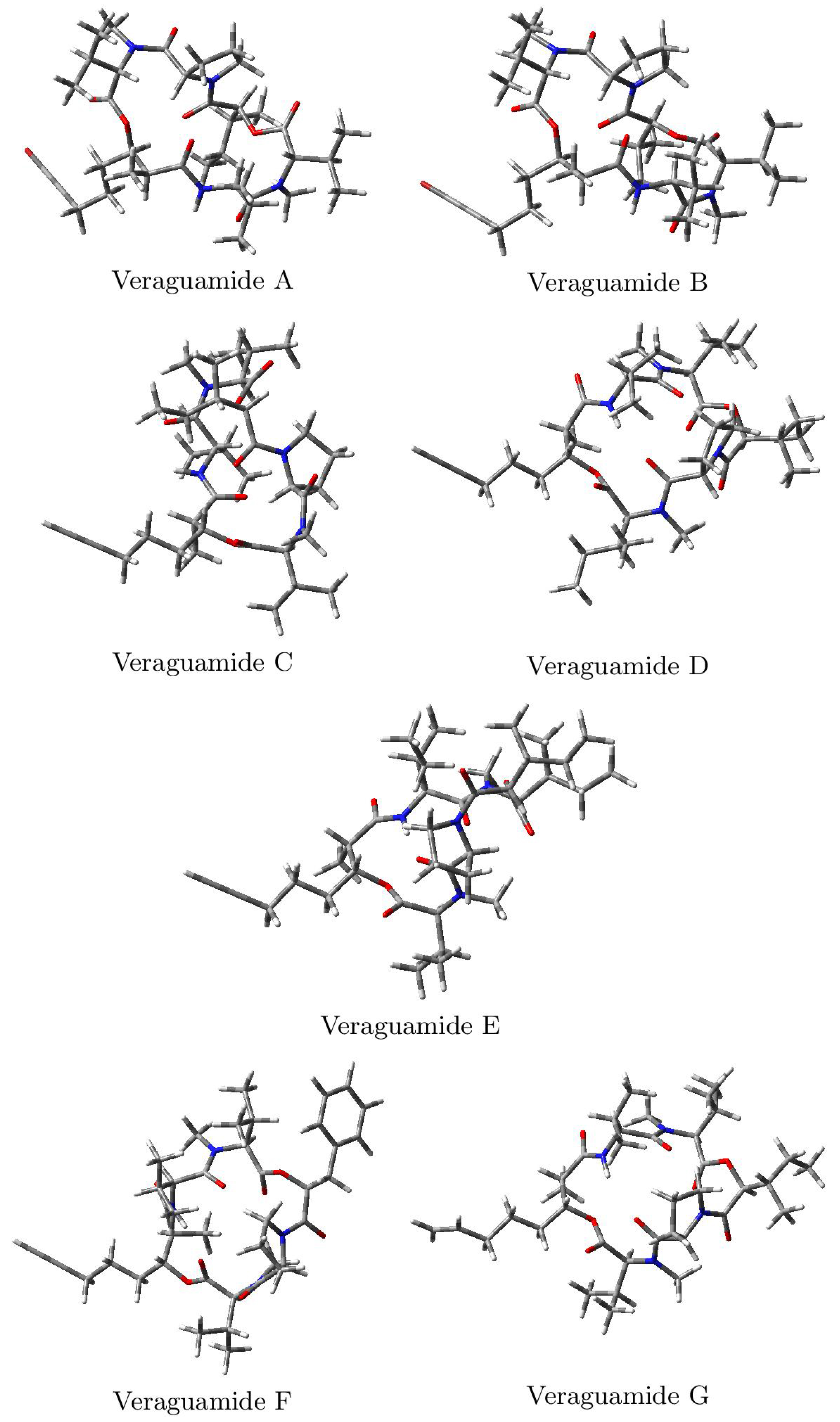
The optimized molecular structures of the Veraguamides A–G marine cyclic hexadepsipeptides computed in accordance with the process shown in the Methodology section are displayed in
Figure 2:
Having verified that the MN12SX/Def2TZVP is the most adequate one for obtaining accurate results for the conceptual DFT global reactivity descriptors, the values for the frontier orbital energies, the HOMO-LUMO gap and the KID indices are presented in
Table 2, while the estimated values for the global reactivity descriptors (including the nucleophilicity N) for the Veraguamides A–G acquired utilizing the mentioned CDFT tool are displayed in
Table 3:
The electronegativity (
) and global hardness (
) are absolute values for the chemical reactivity that have no experimental counterpart. Indeed, they can be estimated by resorting to the experimental vertical ionization energy (I) and vertical electron affinity (A). However, these values are not known for the molecule under study. Going back to the original studies of Robert G Parr and Ralph G Pearson, some kind of classification was done in terms of the HASB principle. This was done only for atoms, ions or very small molecules, for which experimental values for I and A were available at this time. For molecules of the size of the one that we are studying through this research, no standard or experimental values exist. It can only be said something about their global reactivity by comparing with other molecules of the same size. Following this criteria, when comparing with the values of the hardness of some peptides that have been studied recently, it can be said that the veraguamides A–G will be a bit less reactive than those used for comparison because their global hardness values are larger. A different thing can be said about the electrophilicity
and the nucleophilicity (N). The electrophilicity
index involves a compromise between the tendency of an electrophile to acquire extra electron density and its resistance to exchange electron density with the environment [
50]. By considering a group of Diels-Alder reactions and the electrophiles involved in them [
48,
75,
76], a classification of organic compounds as strong, moderate, or marginal electrophiles, that is, an electrophilicity
scale, was established, with
larger than 1.5 eV for the first instance, with
between 0.8 and 1.5 eV for the second case, and
smaller than 0.8 eV for the final case [
48,
75,
76]. By checking
Table 3, it can be said that all the Veraguamides A–G may be regarded as moderate electrophiles. Domingo and his collaborators [
46,
47,
48,
49,
50] have also proposed a nucleophilicity index N through the consideration of the HOMO energy obtained through the KS scheme with an arbitrary shift of the origin taking the molecule of tetracyanoethylene (TCE) as a reference. An analysis of a series of common nucleophilic species participating in polar organic reactions allowed them to establish a further classification of organic molecules as strong nucleophiles with N > 3.0 eV, moderate nucleophiles with 2.0 < N < 3.0 eV and marginal nucleophiles with N < 2.0 eV. As seen in
Table 3, it can be concluded that, with the exception of veraguamide C and veraguamide E, all the other peptides may be considered as moderate nucleophiles.
The global descriptors demonstrate the chemical reactivity of a each molecule in its entirety; therefore, local reactivity descriptors have been designed to assess the differences in the chemical reactivity between the areas inside a molecule. The nucleophilic and electrophilic fukui functions (NFF and EFF) [
25,
26,
27] and the dual descriptor DD [
77,
78,
79,
80,
81,
82] are some of these local reactivity descriptors. They have been defined as: NFF =
, EFF =
and DD =
, establishing links between the electronic densities of the various species as well as between the NFF and EFF. The NFF identifies molecular locations that are more vulnerable to nucleophilic attacks, whereas the EFF identifies regions that are more vulnerable to electrophilic attacks. The reactive locations have been successfully identified using these local reactivity characteristics. However, the dual descriptor DD has been discovered to be capable of describing both nucleophilic and electrophilic locations within a molecule without ambiguity [
82].
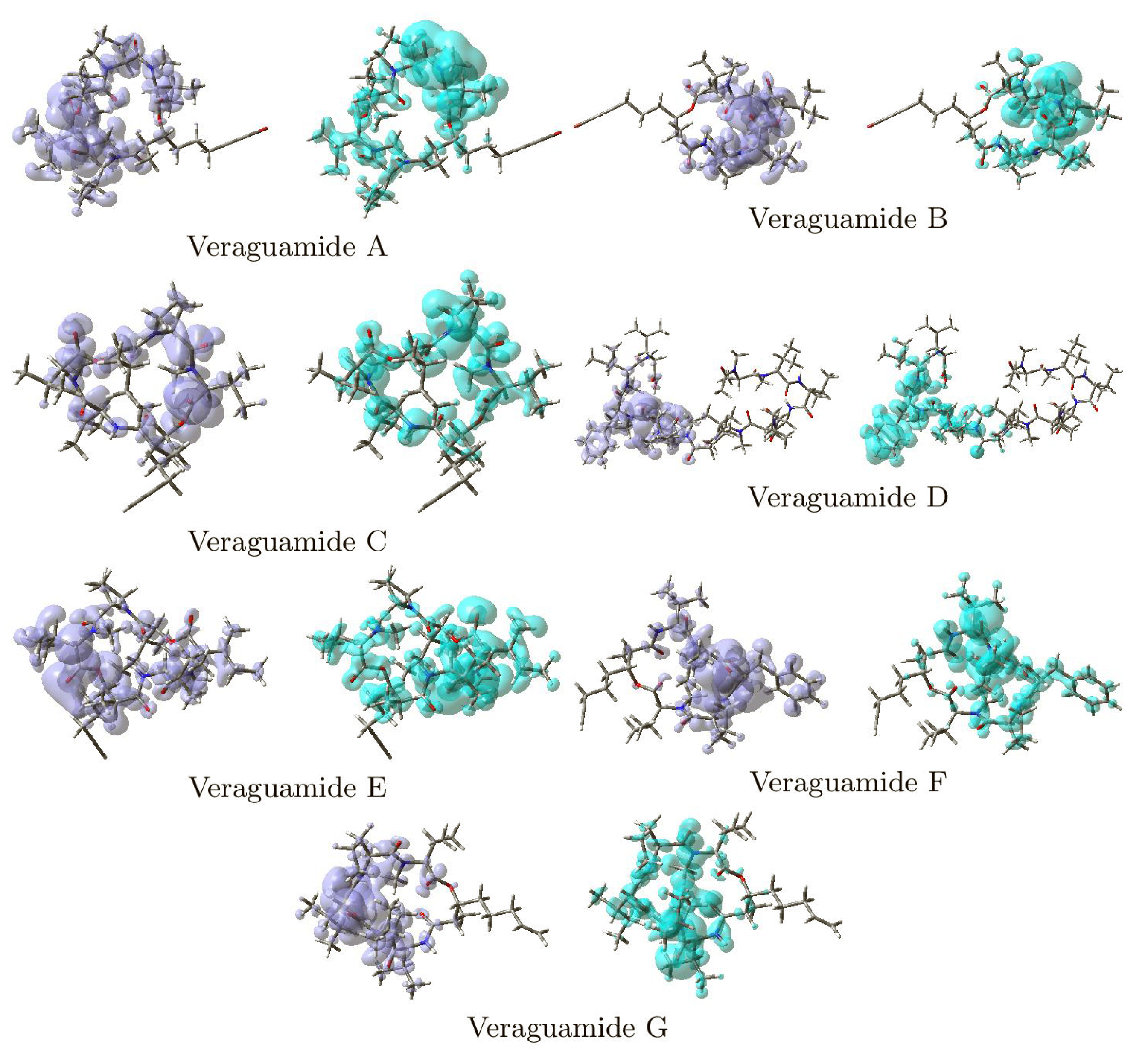
Figure 3 shows graphical sketches of the Dual Descriptor DD for the Veraguamides A–G, highlighting the locations where DD > 0 and DD < 0 for a better understanding of these molecules’ local chemical reactivity.
3.2. Computational Pharmacokinetics and ADMET Report
On the basis of the methodology presented previously, the pKas of the peptides have been estimated following a simple QSAR relationship pKa = 16.3088 − 0.8268
that we have derived during the study of amino acids and small peptides and which has been useful in the study of larger peptides as well as being of interest for the development of advanced glycation end products (AGEs) inhibitors [
83]. These results are reported in
Table 4:
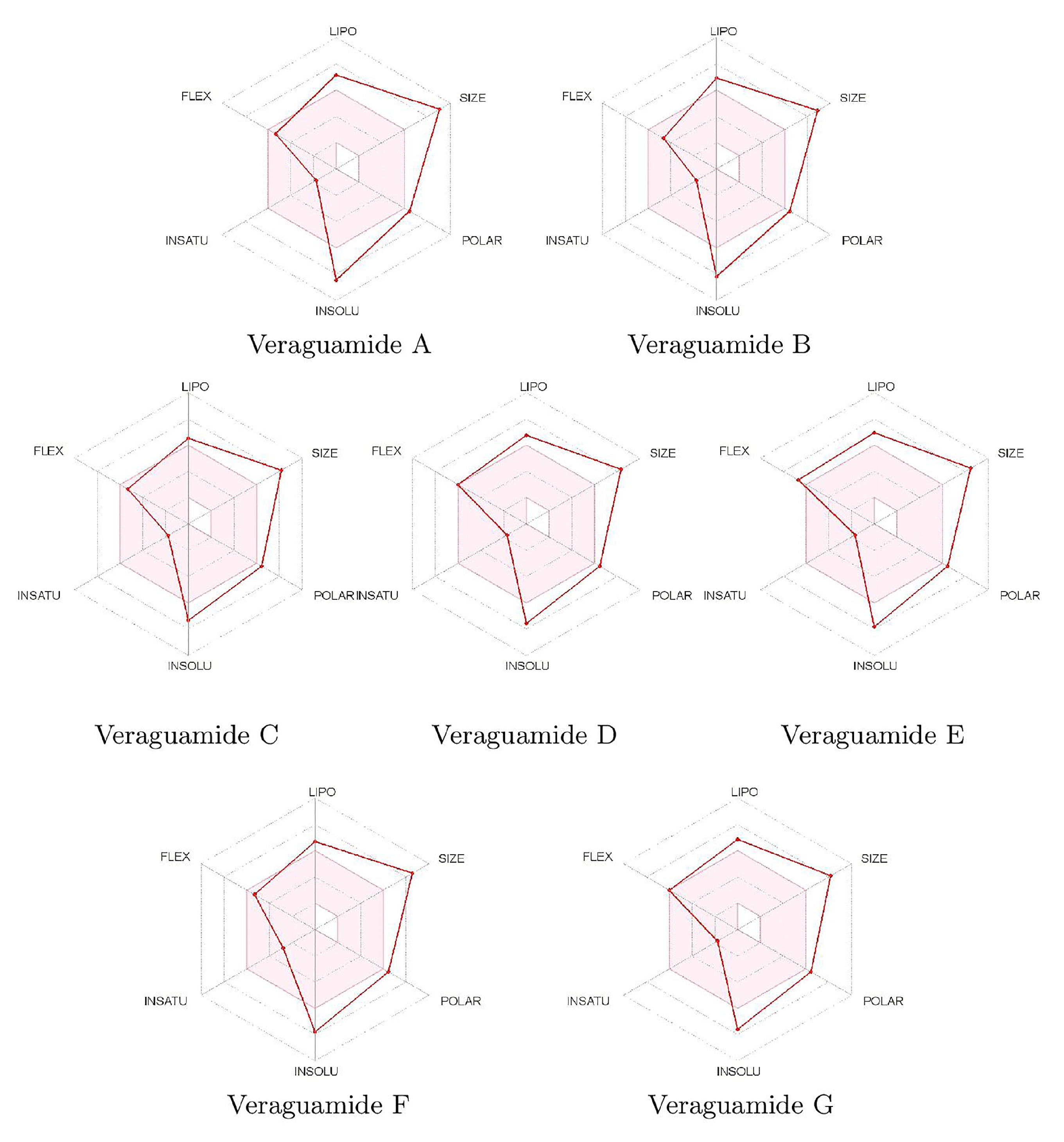
A compact depiction of the characteristics of the molecules related to their bioavailability can be displayed in a graphical mode through the so-called bioavailability radars displayed in
Figure 4 for the veraguamides A–G family of marine cyclic peptides:
It follows that the main difficulties for the veraguamides A–G marine cyclopeptides to be considered as therapeutic drugs of wide bioavailability are those related to their sizes and lack of solubility, and to some extent to their polarities, whose values are somewhat larger than the ideal ones.
The majority of medicinal drugs work by attaching to target protein molecules while at the same time modifying their functions. The Bioactivity Scores, which are a measure of a capacity of the molecules to act or coordinate with distinct receptors, are listed in
Table 5 for the veraguamides A–G.
These bioactivity scores for organic molecules can be interpreted as active (when the bioactivity score is greater than 0), moderately active (when the bioactivity score lies between −5.0 and 0.0) and inactive (when the bioactivity score is lower than −5.0).
The main conclusion from the results of
Table 5 is that the veraguamides A–G will exert their abilities as therapeutic drugs mainly behaving as protease inhibitors, and to a lesser extent, as GPCR ligands and enzyme inhibitors.
The pharmacokinetics of a drug is evaluated through ADMET research, which is an acronym for absorption, distribution, metabolism, excretion, and toxicity. If absorption is unsatisfactory, the distribution and metabolism of the drug would be changed, potentially resulting in nephrotoxicity and neurotoxicity. As a result, ADMET analysis is one of the most important aspects of computational drug design. In addition to the previous conceptual DFT-based computational peptidology and pharmacokinetics results, we are supplementing this study with a report of the computed ADMET features as shown in
Table 6:
It is important to note that all the members of the veraguamides A–G family of cyclopeptides display positive values for the human gastrointestinal absorption (HI) and blood-brain barrier (BBB) permeability and negative values for the AMES toxicity, while the opposite is related to hepatotoxicity. All the peptides will be P-glycoprotein inhibitors (P-gp), being also P-gp substrates. None of the peptides will be inhibitors of the molecules related to cytochrome P450, while all of them will act as substrates of the CYP3A4 variant. Finally, all the cyclic peptides considered here will display a negative result regarding their behavior as hERG inhibitors.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}