1. Introduction
Primary liver cancer, mainly hepatocellular carcinoma (HCC), is the fourth leading cause of worldwide cancer deaths, accounting for an estimated 780,000 deaths annually [
1,
2]. Hepatocellular carcinoma (HCC) is the most prevalent subtype responsible for liver cancer death. Despite advances in diagnosis and treatment, most HCC patients are diagnosed at advanced stages when predicted survival is less than 1 year [
3,
4]. Thus, novel therapeutic agents against HCC are urgently required.
Bioactive compounds derived from natural products are promising candidates for cancer drug development [
5,
6]. In the past four decades, natural anticancer products accounted for approximately 15.4% of new molecular drugs [
7]. Marine organism-produced natural compounds have multifunctional biological activities and provide a new pool of potential cancer treatment options [
8,
9]. Cytarabine, the first marine derivative discovered 50 years ago, was originally isolated from a sponge and was approved by the FDA for the treatment of leukemia in 1969 [
10].
In recent years, bioactive marine alkaloids have been found to be effective in several therapeutic fields such as antibacterial, antioxidant, and anticancer [
11]. Among alkaloids, guanidine is a strong base, which has become a key motif of many clinical drugs such as the famous antidiabetic drug, metformin (dimethyl-biguanide), and the drug for treating peptic ulcers, cimetidine. In terms of anticancer effects, previous in vitro studies have shown that guanidine marine alkaloids have potential anticancer properties and cytotoxic activities [
12]. Crambescidin-816 is a pentacyclic guanidine alkaloid isolated from
Crambe crambe (oyster sponge), which could reduce the cell viability of liver cancer cells in experiments [
13].
Marine sponges of the
Agelas genus were found to be prolific producers of bromopyrrole derivatives and terpenoid alkaloids. The first isolated derivative and a major sesquiterpene alkaloid possessing sulfone and guanidine functional groups, agelasidine A, was discovered and reported by Nakamura et al. in 1983 from
Agelas sp. This organism was identified as
Agelas nakamurai in a related article published in 1985, with the value of specific optical rotation being +19.1 [
14,
15]. Subsequently, the same metabolite referred to as agelasidine A or, more specifically, (+)-agelasidine A was again isolated from a sponge of the same genus of an unknown species in 1984 by Capon et al. [
16]. (+)-Agelasidine A has been found to display antispasmodic and antibacterial activities [
16]. In 2006, the optical isomer (−)-agelasidine A, with the value of specific optical rotation being −12.2, was discovered for the first time from the sponge
A. clathrodes by Medeiros et al. [
17]. However, so far, there are still very few studies focusing on the biological activity of (−)-agelasidine A, except for previous studies that proved its antibacterial effect [
17]. In addition, (−)-agelasidine A challenge against cancer has not been explored in the currently available literature. To expand knowledge of this compound, we determined the cytotoxic effects of (−)-agelasidine A isolated from
Agelas nakamurai in human Hep3B and HepG2 liver cancer cell lines. In this paper, we also explored how (−)-agelasidine A induced cancer cell death.
3. Discussion
HCC is one of the most frequently diagnosed malignancies, and the vast majority of patients are diagnosed too late to receive surgeries. In this way, many of the patients need alternative options such as chemotherapies and target therapies. Sorafenib and lenvatinib, tyrosine kinase inhibitors that target angiogenesis in cancer cells, are the currently licensed treatment options for metastatic HCC, HCC ineligible for surgeries, and HCC cases unable to undergo focal therapies. However, treatment effects are sometimes unreliable, and the average recovery time of treated patients is around 8 months and 4 months with lenvatinib and sorafenib treatment, respectively [
18]. Exploration of anticancer drugs that are safer and more efficient is essential for the progress of modern medicine.
Ocean invertebrates offer a diverse source of metabolites with anticancer activities, and many of them are licensed for cancer treatment [
5,
19]. (−)-Agelasidine A, an alkaloid extracted from
Agelas nakamurai, has been shown to have antifungal abilities [
17], yet its anticancer abilities have not been investigated. In this study, through in vitro experiments, the anticancer abilities of (−)-agelasidine A were demonstrated. On the basis of our current understanding, (−)-agelasidine A induces ER stress followed by apoptosis to produce anticancer effects.
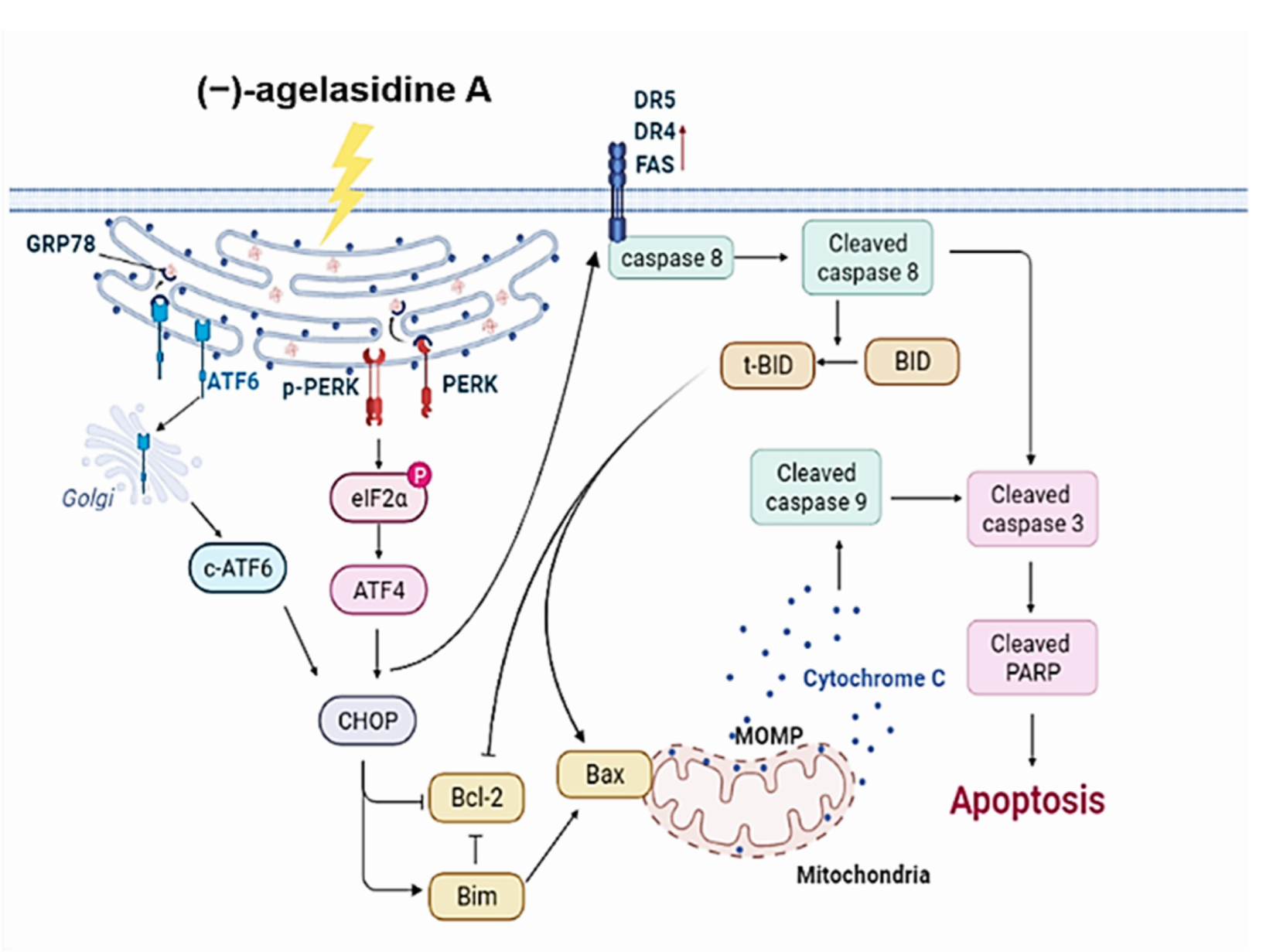
Figure 9 demonstrates the proposed mechanism of (−)-agelasidine A-driven apoptosis in Hep3B cell lines.
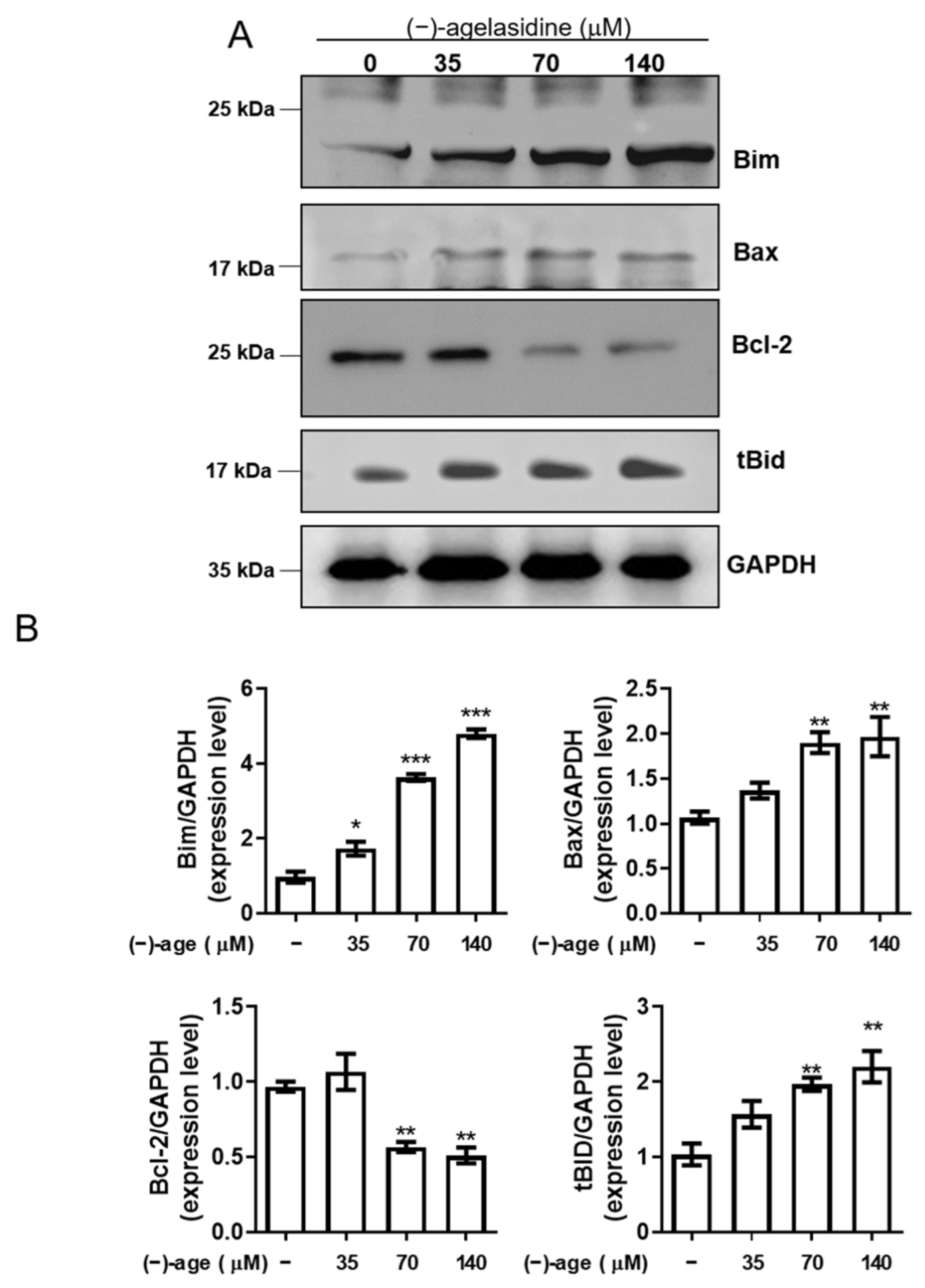
Apoptosis is a process where dysfunctional or damaged cells undergo programmed cell death, and it is an essential mechanism via which organisms maintain homeostasis [
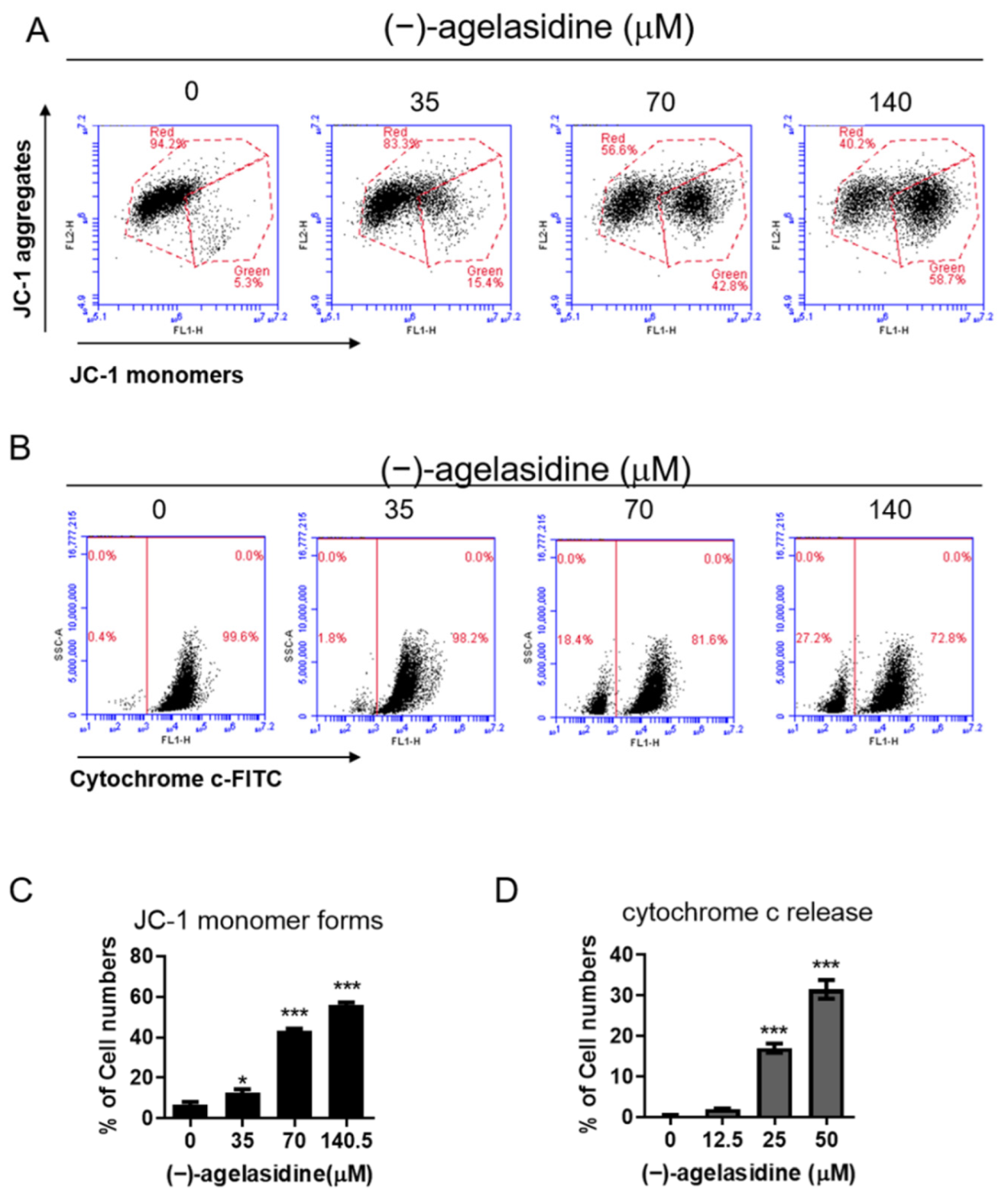
20]. Apoptosis is also an essential pathway responsible for eradication of cancerous cells that arise in the body. In this study, Hep3B cellular apoptosis was promoted after introducing (−)-agelasidine A. Considering that there are intrinsic and extrinsic pathways in the regulation of cellular apoptosis, we examined the proapoptotic function of (−)-agelasidine A according to signals in each pathway (
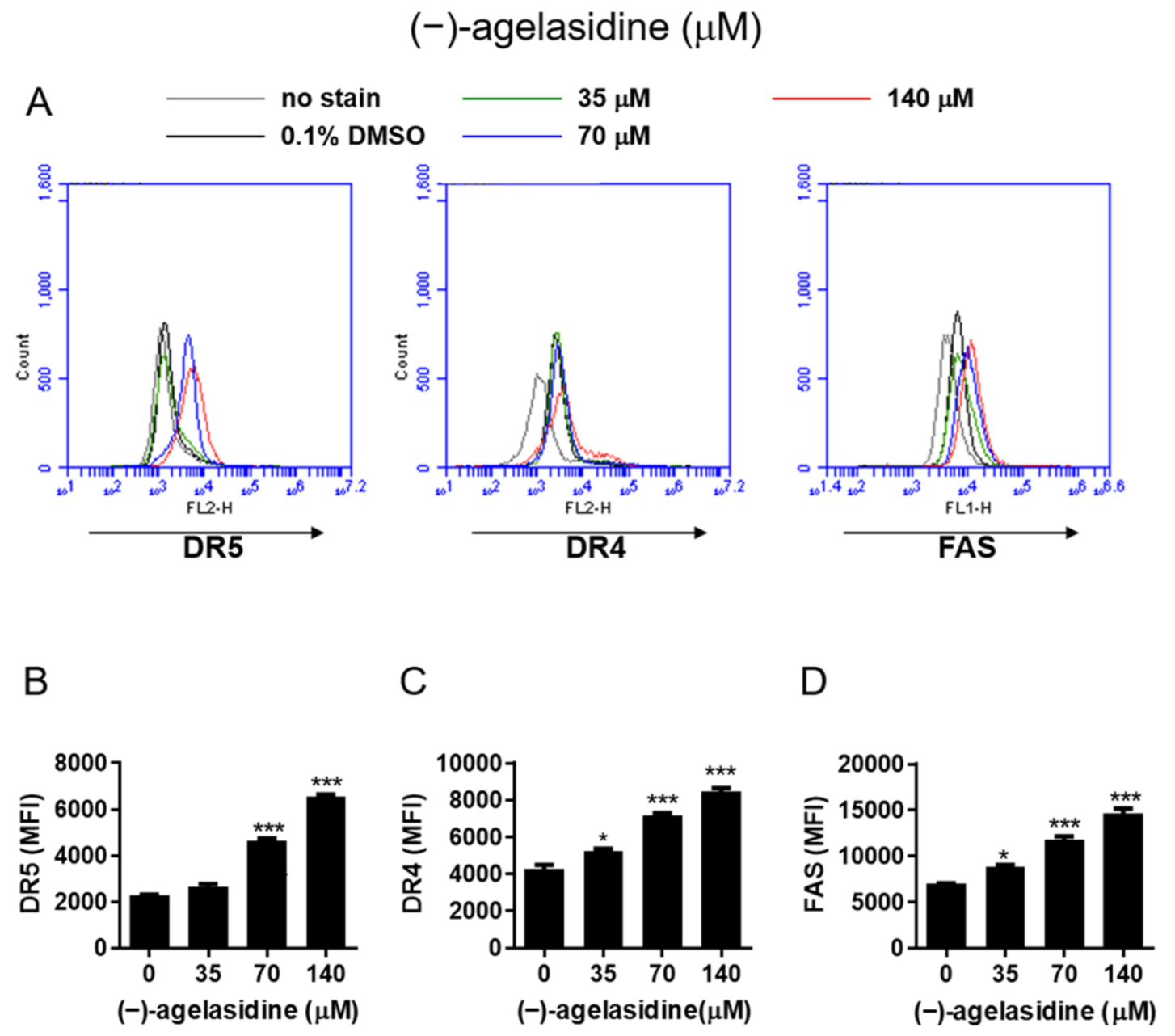
Figure 9). The intrinsic pathway is initiated by the loss of MMP and mitochondrial membrane perturbation. In this process, complexes of antiapoptotic Bcl-2 and Bad stimulate the release of proapoptotic Bax, while permeability of the mitochondria is altered by Bim. This causes the release of cytochrome c from mitochondria to the cytoplasm. Subsequently, executioner caspase-3/PARP is activated by apoptotic bodies. The extrinsic pathway is driven by the formation of death-inducing signaling complex (DISC) and activation of caspase-8. After proapoptotic tBID is processed by caspase-8, it binds to antiapoptotic Bcl-2 and upregulates the release and activity of proapoptotic Bax. Consequently, the extrinsic pathway can be connected to the intrinsic pathway. In our experiment, (−)-agelasidine A could promote apoptosis via the intrinsic pathway, including inducing proapoptotic Bax and Bim, reducing antiapoptotic Bcl-2, altering MMP, facilitating release of cytochrome c, and enhancing executioner caspase-9. By increasing death receptors (DRs) such as DR4, DR5. and FAS, (−)-agelasidine A could also upregulate caspase-8 and tBID and, consequently, stimulate both extrinsic and intrinsic pathways (
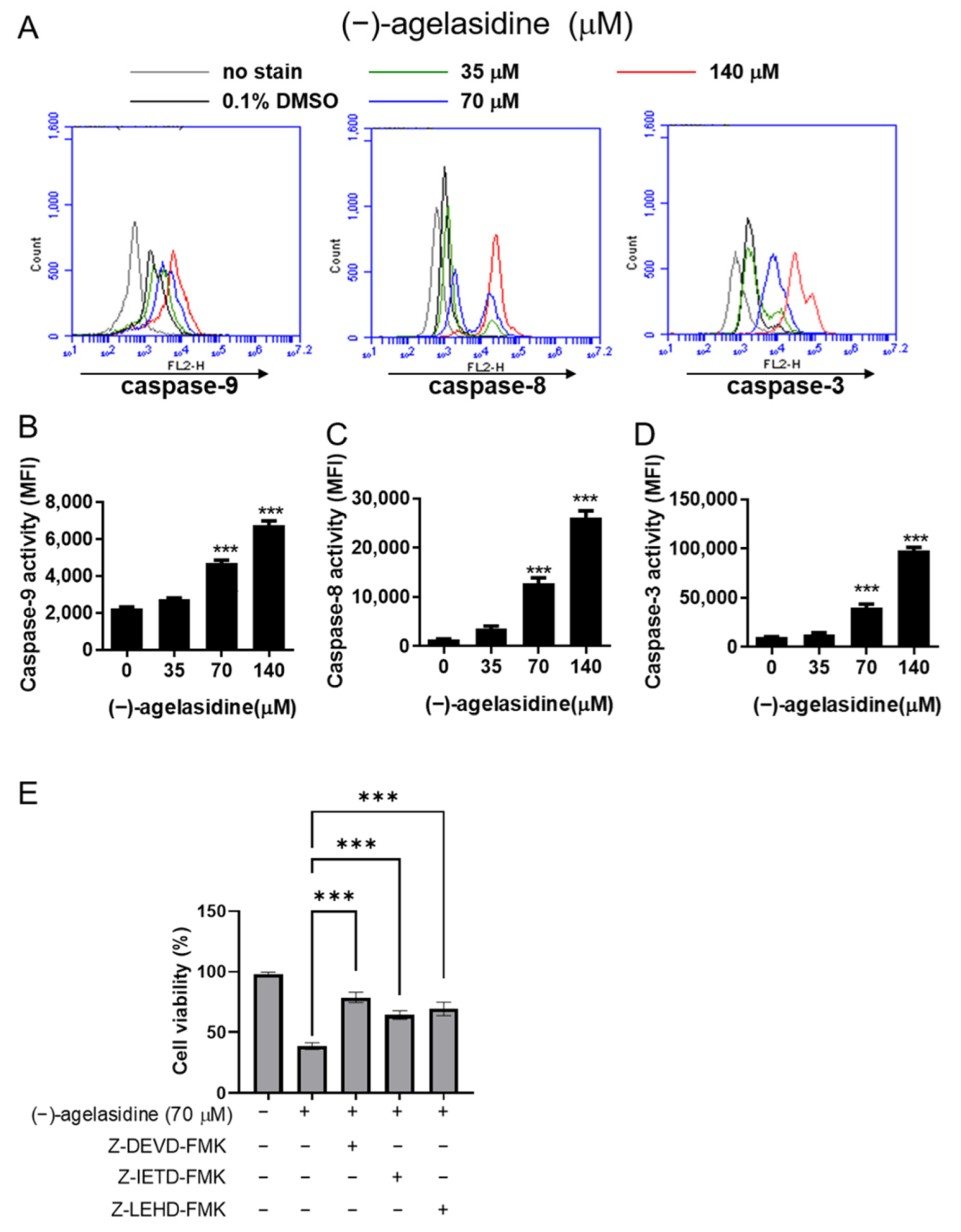
Figure 6). Our inhibitory experiment with caspase-3, -8, and -9 inhibitors also indicated the multiple possible mechanisms of action of (−)-agelasidine A in inducing apoptosis in cancerous cells regardless of intrinsic and extrinsic pathways.
The ER is essential in maintaining cellular homeostasis and inducing cytotoxic cellular death. Its function can be disrupted by different pathophysiological conditions that cause the accumulation of unfolded proteins. This results in ER stress and leads to activation of the unfolded protein response (UPR). The UPR can then initiate cellular apoptosis. In mammal cells, UPR caused by ER stress is regulated by three ER transmembrane proteins, which are ATF6, PERK, and IRE1α [
21,
22]. Emerging evidence shows that natural compounds and their derivatives have the potential to treat cancers, metabolic diseases, cardiovascular diseases, and neurodegenerative disorders by regulating ER stress-related pathways [
23,
24,
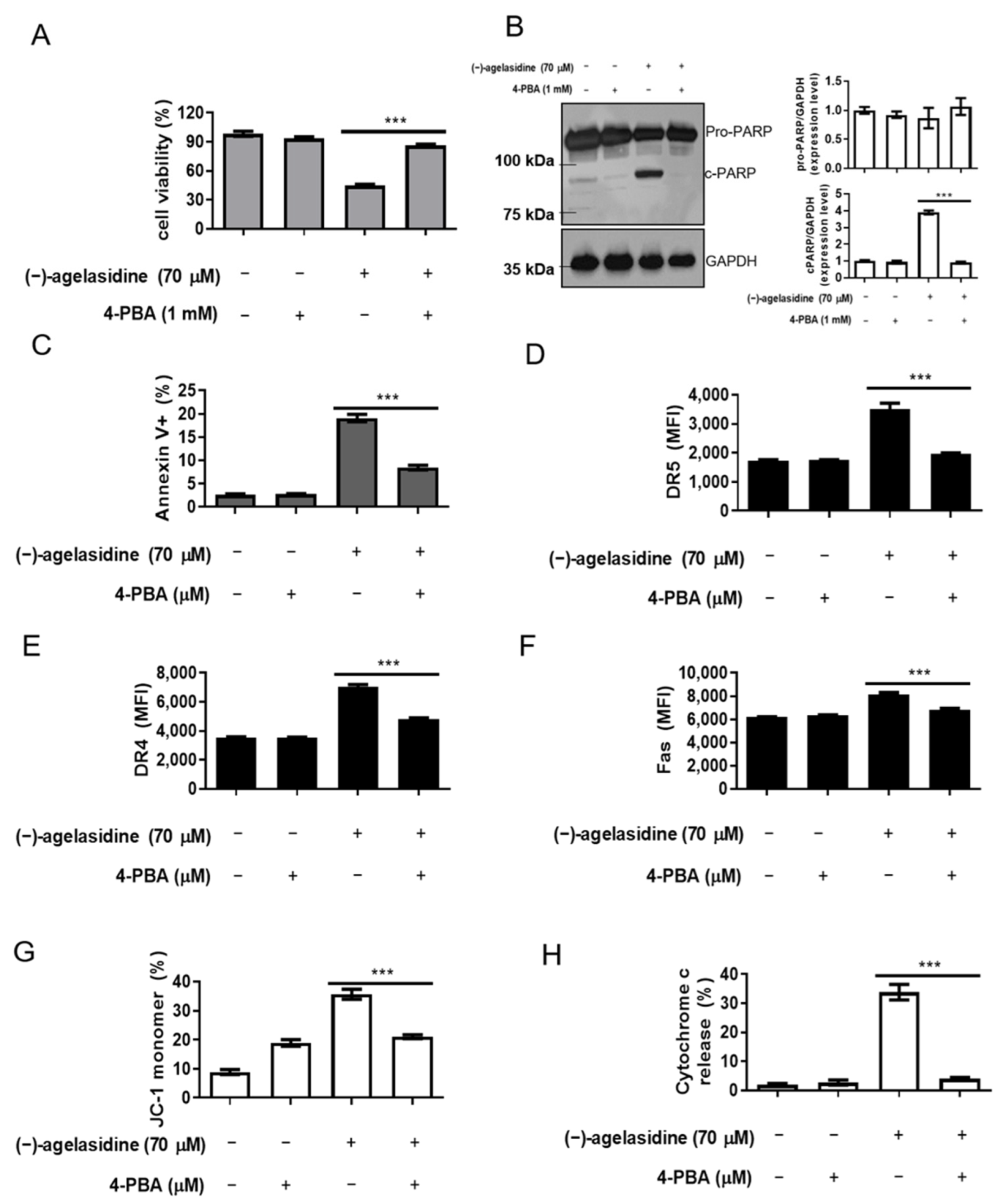
25]. In our study, (−)-agelasidine A could induce ER stress in dose-dependent and time-dependent fashions. It also demonstrated that ER-stress-related proteins such as GRP78, p-PERK, p-eIF2α, ATF4, c-ATF6, and CHOP are all upregulated during treatment. Through inhibitor-driven CHOP suppression, our studies succeeded in reducing ER stress-induced cellular apoptosis. In our experiment, adding 4-PBA to inhibit ER stress could suppress DRs, as well as accomplish MMP loss and release of cytochrome c. Decreased cellular apoptosis was also noted. These results suggest that (−)-agelasidine A initiated cellular apoptosis through enhancing ER stress in HCC.
In the past 20 years, marine invertebrates have been unveiled as important, prolific sources of bioactive secondary metabolites. Many of these compounds have shown potential as anticancer medications worth further research [
26]. However, although the efficacy of these marine natural compounds in fighting diseases has been distinguished, the sources are limited, and only finite amounts of natural products can be produced. Therefore, difficulties arise when trying to acquire a sufficient supply for drug discovery and development. Moreover, in the long run, collecting bioactive agents from marine invertebrates is not practical for clinical trials and mass production for pharmaceutical applications. Accordingly, the production of these potential natural pharmaceuticals via artificial methods is a more feasible means of developing such medications. Another potential approach to overcome the limited quantities of compounds from natural sources is through aquaculture of marine organisms by optimizing their growth environment in cultured tanks or pools [
27,
28]. A final potentially useful way to provide these medicinally attractive compounds is through chemical synthesis. With great advances in chemical synthesis, steady and controllable manufacturing processes could effectively increase the production of bioactive natural compounds and allow us to improve bioavailability by modifying their structures [
29,
30]. For the supply of (−)-agelasidine A for research and drug development, this barrier is less significant since 1227 mg of this compound could be isolated from 5 kg of the sponge
A. clathrodes [
17]. Thus, sufficient amounts of (−)-agelasidine A can be provided for medicinal application if aquaculture of this sponge can be achieved in the future. Furthermore, racemic agelasidine A was synthesized in 1992 [
31] from commercially available farnesol via an efficient three-step synthesis. In 2019, optically active compound (−)-agelasidine A was successfully and efficiently synthesized by catalytic hydrothionation of β-farnesene [
32]. It was also synthesized by regioselective and enantioselective synthesis based on palladium-catalyzed allylic sulfonylation from the tertiary allylic carbonate of farnesonol [
33]. Thus, once (−)-agelasidine A is proven to be an effective agent for inhibition of in vivo HCC, this compound can be readily provided for further clinical trials of HCC treatment.
4. Material and Methods
4.1. Cell Culture
The HepG2 and Hep3B cancerous cell lines were purchased from the Food Industry Research and Development Institute (Hsinchu City, Taiwan). All cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/mL penicillin, and 100 g/mL streptomycin. Cells were incubated at 37 °C in a humidified atmosphere containing 5%/95% of CO2/air. Cell culture reagents were purchased from Invitrogen Life Technologies (Carlsbad, CA, USA).
4.2. Chemicals
The frozen bodies of
Agelas nakamurai, collected from the coast of Orchid Island (Taiwan) in 2008 and identified by Dr. Meng-Chen Yu at Biodiversity Research Center, Academia Sinica, (Taipei, Taiwan), were sliced and extracted exhaustively with ethyl acetate (EtOAc). The combined EtOAc extract was evaporated under reduced pressure to give a crude residue, which was subjected to silica gel column chromatography, eluting with EtOAc in
n-hexane (0–100%, stepwise) and then with MeOH in acetone (0–100%, stepwise), to yield 19 fractions (F1–F19). Fraction 13 was chromatographed by elution with MeOH–acetone (3:1) to yield a partially pure compound (−)-agelasidine A. This compound was purified by chromatography over a reversed-phase C18 column using MeOH–H2O (4:1) as the eluent to afford pure (−)-agelasidine A, which was subsequently confirmed by NMR and specific optical rotation, both of which appeared to be identical with data reported previously ([α]
−24.9 and
Figure S2 [
17]).
4.3. Cell Viability Assay
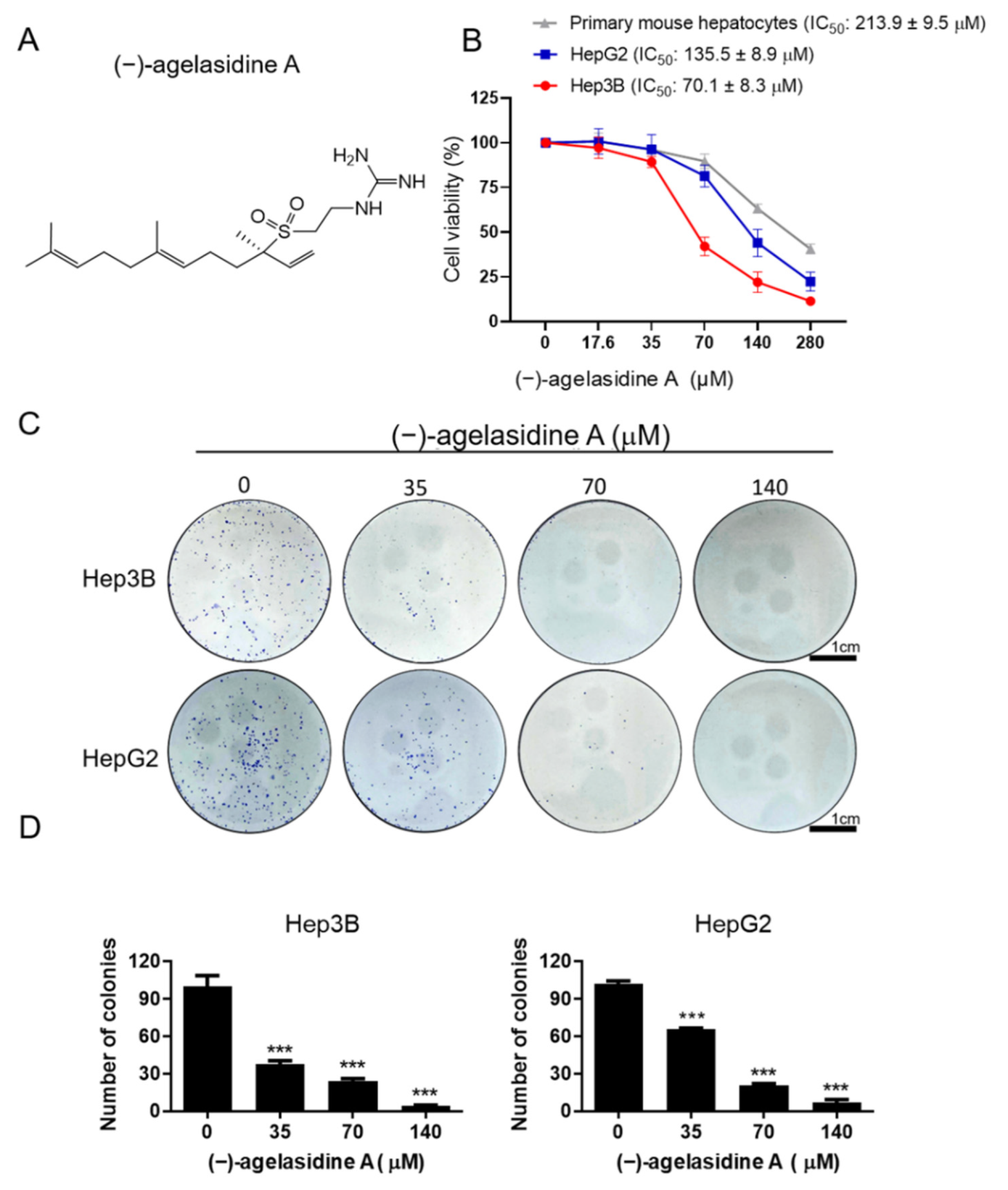
Cancerous cells, Hep3B and HepG2, were seeded into 24-well plates at 2 × 104 cells/well and treated with various concentrations of (−)-agelasidine A or 0.1% DMSO as a control vehicle for 24 h. Following incubation, 10 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich, St. Louis, MO, USA) solution (0.5 mg/mL final concentration) was added to each well. The supernatant was aspirated after 4 h, and then 600 μL of DMSO was added to dissolve the formazan crystals. The absorbance was examined at 540 nm using a microplate reader (TECAN, Durham, NC, USA). Data were shown as the percentage absorbance of (−)-agelasidine A-treated cells relative to DMSO-treated cells. The 50% inhibitory concentration (IC50) values were calculated using GraphPad software for semi-log curve fitting with regression analysis. This cell viability assay was also performed to examine the involvement of the caspase pathway or ER stress in the (−)-agelasidine A-mediated cytotoxicity. Briefly, Hep3B cells were incubated overnight and then pretreated with 1 mM 4-PBA (Cayman Chemical Company, Ann Arbo, MI, USA), caspase-3 inhibitor (Z-DEVD-FMK, 10 µM, R&D Systems, Minneapolis, MN, USA), caspase-8 inhibitor (Z-IETD-FMK, 20 µM, BioVision, San Francisco, CA, USA), or caspase-9 inhibitor (Z-LEHD-FMK, 20 µM, BioVision, Milpitas, CA, USA) for 2 h. Cells were then treated with 70 μM (−)-agelasidine A for an additional 24 h prior to MTT assay.
Normal hepatocytes were also isolated by in situ retrograde collagenase perfusion as described previously [
34] and tested for cytotoxicity of (−)-agelasidine A with MTT assay. Briefly, hepatocytes were dissociated from the collagenase-digested liver after PBS perfusion and filtered through a gauze. The cell suspension was then fractionated by Percoll density centrifugation (2500 rpm for 5 min at 4 °C) to purify hepatocytes, followed by incubating at 37 °C under an atmosphere of 95% air–5% CO
2 and testing via MTT assay.
4.4. Colony-Forming Assays
Colony formation assays were carried out to test the effect of (−)-agelasidine A on the clonogenicity of HepG2 and Hep3B cells. Briefly, cells were seeded into six-well plates at 500 cells/well and incubated for 24 h. The cells were then treated with different concentrations of (−)-agelasidine A (35, 70, and 140 μM) for 1 week to allow colonies to form. Crystal violet (2%) (Sigma-Aldrich, St. Louis, MO, USA) was used to stain colonies, and the number of colonies in each well was counted under an inverted microscope (Olympus, Tokyo, Japan).
4.5. Analysis of Cell Apoptosis by Flow Cytometry
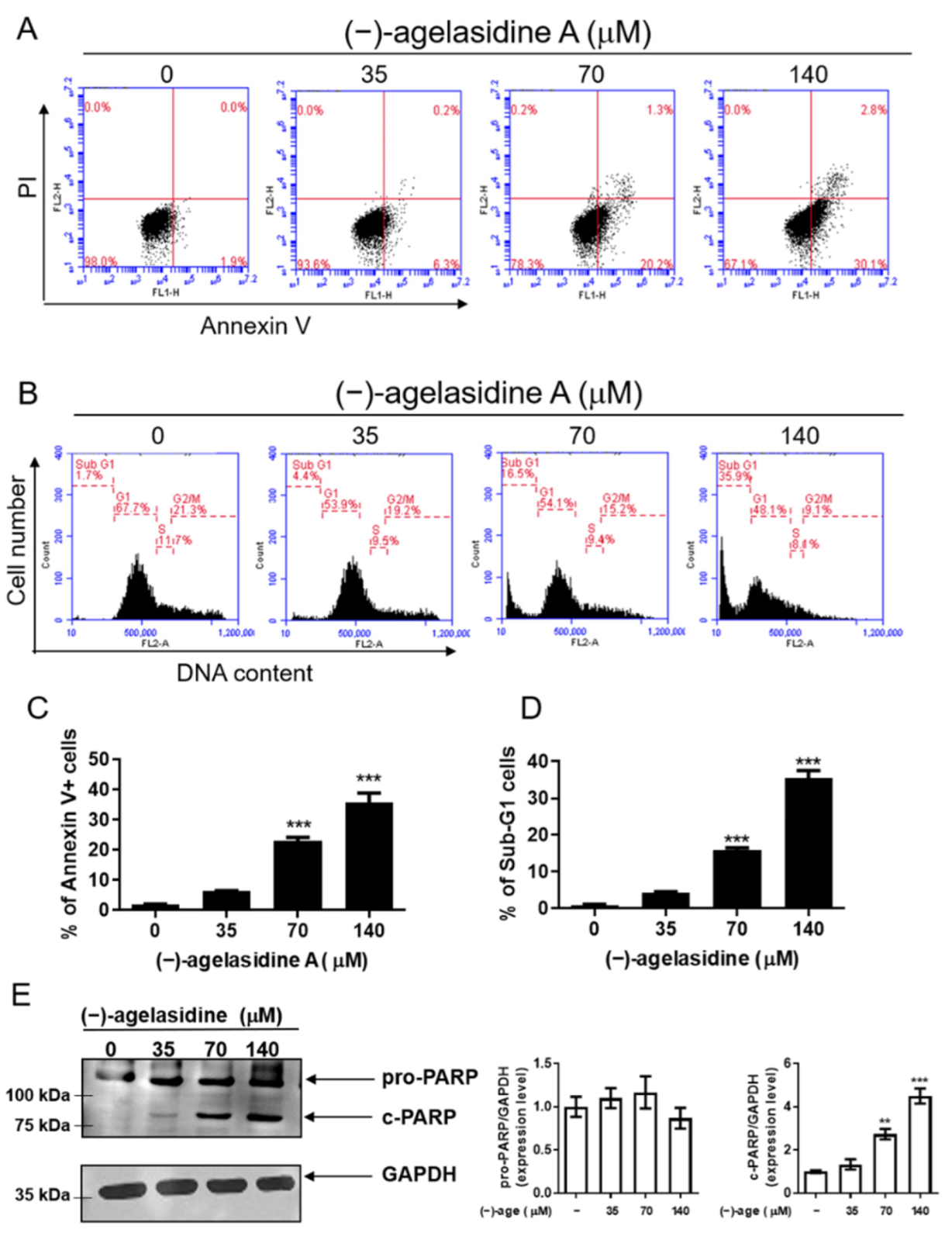
The apoptosis assay was performed using an annexin V/FITC apoptosis detection kit (BD Biosciences, Franklin Lakes, NJ, USA). The Hep3B cells (2 × 105/well) were seeded in six-well plates and incubated overnight. After treatment with (−)-agelasidine A (0, 35, 70, or 140 µM) for 24 h, cells were harvested and washed in PBS. Cells were incubated with 5 μL of annexin V/FITC (20 μg/mL) and 5 μL of propidium iodide (PI) (50 μg/mL) at room temperature for 10 min in the dark. Apoptotic cells were detected using an AccuriTM C5 cytometer (BD Biosciences, Franklin Lakes, NJ, USA), and the data were analyzed using BD Accuri C6 Software version 1.0.264.21 (BD Biosciences, Ann Arbor, MI, USA).
4.6. Determination of DNA Content by Flow Cytometry
Hep3B cells were seeded into six-well plates at a density of 2 × 105 cells/well and treated with (−)-agelasidine A (35, 70, and 140 μM) for 24 h. The cells were harvested and fixed with 70% ethanol overnight at −20 °C. Subsequently, cells were stained with PI/RNase staining buffer (BD Biosciences, San Diego, CA, USA) in PBS for 30 min. DNA content was analyzed using an Accuri C5 cytometry (BD Biosciences, Ann Arbor, MI, USA).
4.7. Western Blot Analysis
Cells were seeded into six-well plates at a density of 2 × 105 cells/well and treated with indicated concentrations of (−)-agelasidine A for 12 h (for ER stress molecules) or 24 h. The cells were collected, and cell pellets were lysed in RIPA buffer containing 1% protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA) and phosphatase inhibitor (Roche, Mannheim, Germany). The BCA protein assay (Thermo Fisher Scientific, Waltham, MA, USA) was used for quantitation of total protein. An equal amount of protein was immobilized by 5–12% sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (Merck Millipore, Billerica, MA, USA). The membranes were then blocked in BlockPRO Protein-Free Blocking Buffer (Visual protein, Taipei City, Taiwan) for 1 h, followed by incubation with anti-PARP (Polyconal, 1:1000; Cell Signaling Technology, Danvers, MA, USA), anti-Bim (clone C34C5, 1:1000; Cell Signaling Technology,) anti-Bax (clone 2D2, 1:3000; BioLegend, San Diego, CA, USA), anti-Bcl-2 (clone 100, 1:2000; BioLegend, San Diego, USA), anti-Bid (Clone 2002, 1:1000; Cell Signaling Technology, Danvers, MA, USA), anti-GRP78 (Polyclonal, 1:10,000; GeneTex Inc., Irvine, CA, USA), anti-phospho-PERK(Thr 980) (Polyclonal, 1: 1000; Bioss, Woburn, MA, USA), anti-PERK (Clone D11A8, 1:1000; Cell Signaling Technology, Danvers, MA, USA), anti-Phospho-eIF2α (Clone 119A11, 1:1000; Cell Signaling Technology, Danvers, MA, USA), anti- eIF2 (polyconal, 1:1000; Cell Signaling Technology, Danvers, MA, USA), ATF4 (clone D4B8, 1:1000; Cell Signaling Technology, Danvers, MA, USA); ATF6 (clone W17028A, 1:1000; BioLegend, San Diego, CA, USA), anti-CHOP (clone L63F7, 1:1000; Cell Signaling Technology, Danvers, MA, USA), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (clone 6C5, Abcam, Cambridge, MA, USA) antibodies at 4 °C overnight. The next day, the membrane was incubated with the horseradish peroxidase (HRP)-conjugated corresponding secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) at 4 °C overnight. After washing with TBST, the immunoreactive band was detected using LumiFlash Ultima Chemiluminescent substrate in an HRP system (Visual protein, Taipei City, Taiwan; LF08–500) and imaged using a Hansor Luminescence Image System (Taichung, Taiwan). The band densities were analyzed using the ImageJ 1.47 program from the National Institute of Health (NIH) (Bethesda, MD, USA).
4.8. Caspase Activity Assay by Flow Cytometry
The cells were seeded into six-well plates at a density of 2 × 105 cells/well and treated with (−)-agelasidine A (35, 70, and 140 μM) for 24 h. Caspase-3, -8, and -9 activities were measured using the appropriate CaspGLOW fluorescein active caspase staining kits (Biovision, Milpitas, CA, USA) according to the manufacturer’s protocol and measured by flow cytometry on an Accuri C5 cytometry (BD Biosciences, Ann Arbor, MI, USA).
4.9. Measurement of Mitochondrial Membrane Potential
Mitochondrial membrane potential (MMP) was detected using JC-1 dye (Invitrogen Life Technologies, Carlsbad, CA, USA). Briefly, Hep3B cells were treated with (−)-agelasidine A (35, 70, and 140 μM) for 24 h, collected, and washed with PBS. Then, the cells were stained with JC-1 fluorescent dye at 37 °C for 20 min in the dark. After incubation, the level of mitochondrial membrane potential (ΔΨm) was determined using an Accuri C5 cytometer (BD Biosciences, Ann Arbor, MI, USA). JC-1 monomers and J-aggregates were detected in the FL1 and FL2 channels, respectively, whereby variations in the red/green fluorescence intensity ratio reflected changes in the mitochondrial membrane potential.
4.10. In Vitro Assay for Cytochrome c Release from Mitochondria by Flow Cytometry
The release of cytochrome c from the mitochondria to the cytosol was detected by FITC–anti-cytochrome c antibody (Clone 6H2.B4, 1:1000; BioLegend, San Diego, CA, USA). Briefly, cells were seeded into six-well plates (2 × 105 cells/well) and treated with the indicated concentrations of (−)-agelasidine A for 24 h. Subsequently, cells were permeabilized with 100 µL of digitonin lysis buffer (50 µg/mL digitonin and 100 mM KCl in 1× PBS) for 5 min on ice and then fixed in 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA) in PBS for 20 min at RT. Cells were stained with FITC–anti-cytochrome c antibody in 0.5 mL of labeling buffer (2% BSA in 0.05% Triton X-100 PBS) at 4 °C for 35 min, and then detected for cytochrome c-positive cells by Accuri C5 cytometry (BD Biosciences, Ann Arbor, MI, USA).
4.11. Statistical Analysis
All data were presented as the means ± standard deviation (SD), and two-tailed Student’s t-tests were used in examining the null hypothesis of two unpaired independent samples. One-way ANOVA was performed to compare different groups using GraphPad Prism Version 6.0 (San Diego, CA, USA). The acceptable type 1 error was considered to be 5%.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








