

Xestospongia muta Fraction-7 and Linoleic Acid: Effects on SR-BI Gene Expression and HDL Cholesterol Uptake


 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Cytotoxicity of Diethyl Ether Fractions of X. muta
2.2. SR-BI Transcriptional Activation by Diethyl Ether Fractions of X. muta
2.3. GC-MS of Fraction-7 of X. muta
2.4. SR-BI Transcriptional Activation by Potential Fatty Acids
2.5. Isolation and Quantification of Total RNA
2.6. Real-Time PCR and SR-BI Gene Expression
2.6.1. Optimization of Semi-Quantitative Rt-PCR Reaction
2.6.2. Semi-Quantitative Rt-PCR Amplification Efficiency
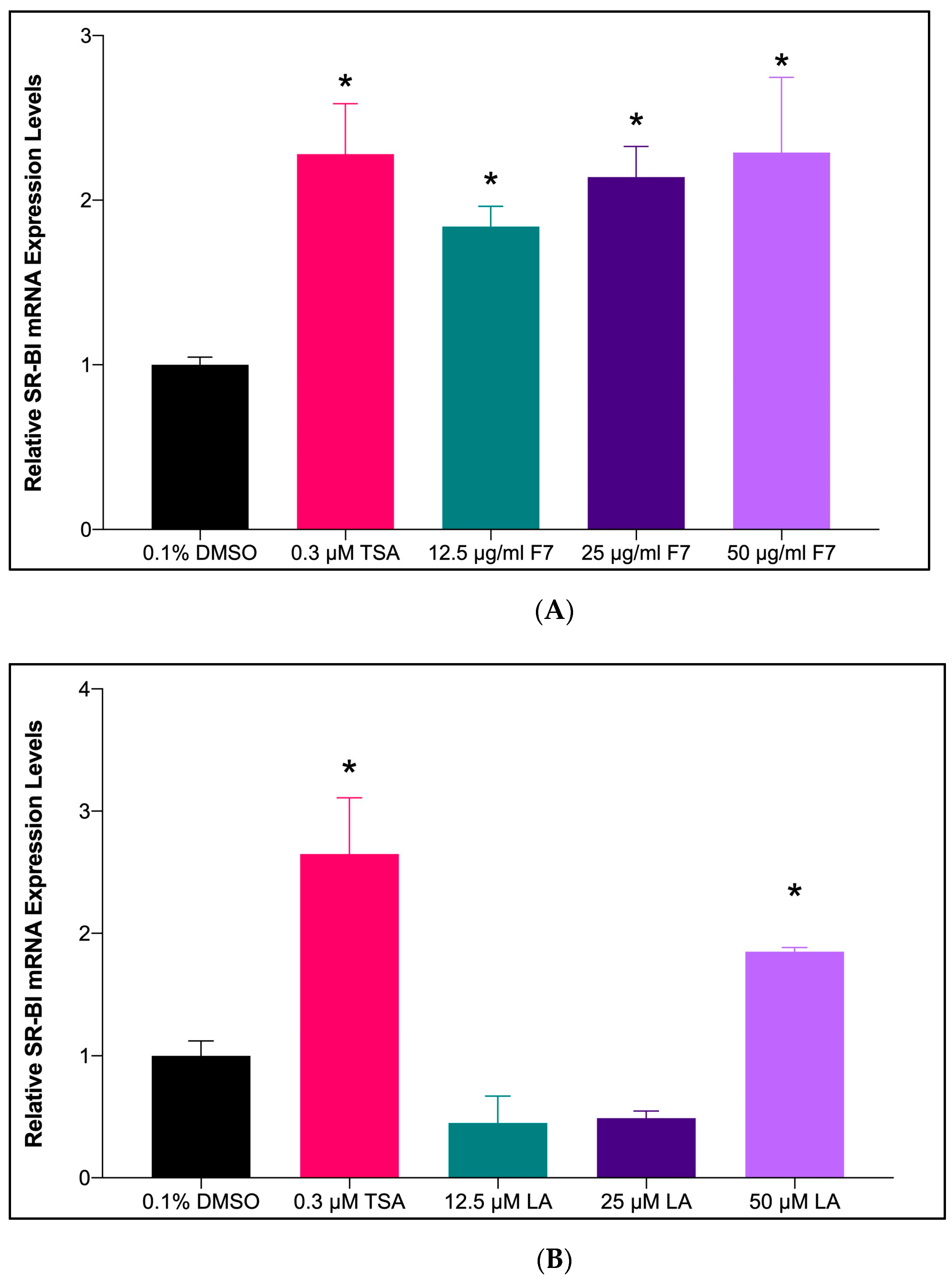
2.6.3. mRNA Expression of SR-BI Gene upon Treatment with Fraction-7 and LA
2.6.4. Time Lapse for SR-BI Expression of HepG2 Cells Treated with Fraction-7 and LA
2.7. HDL-C Uptake and Protein Cellular Localization
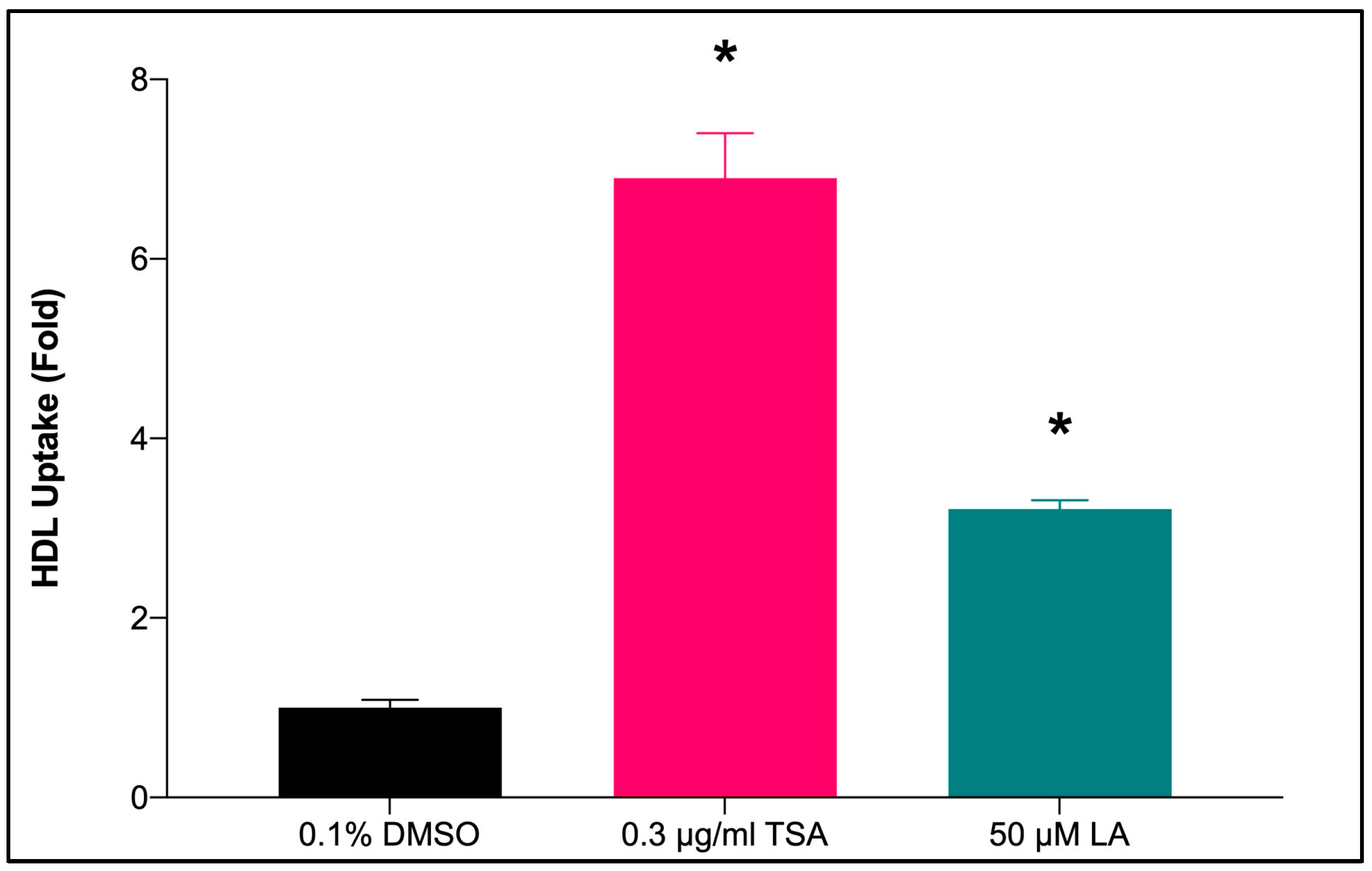
2.7.1. HDL-C Uptake by HepG2 Cells Treated with LA
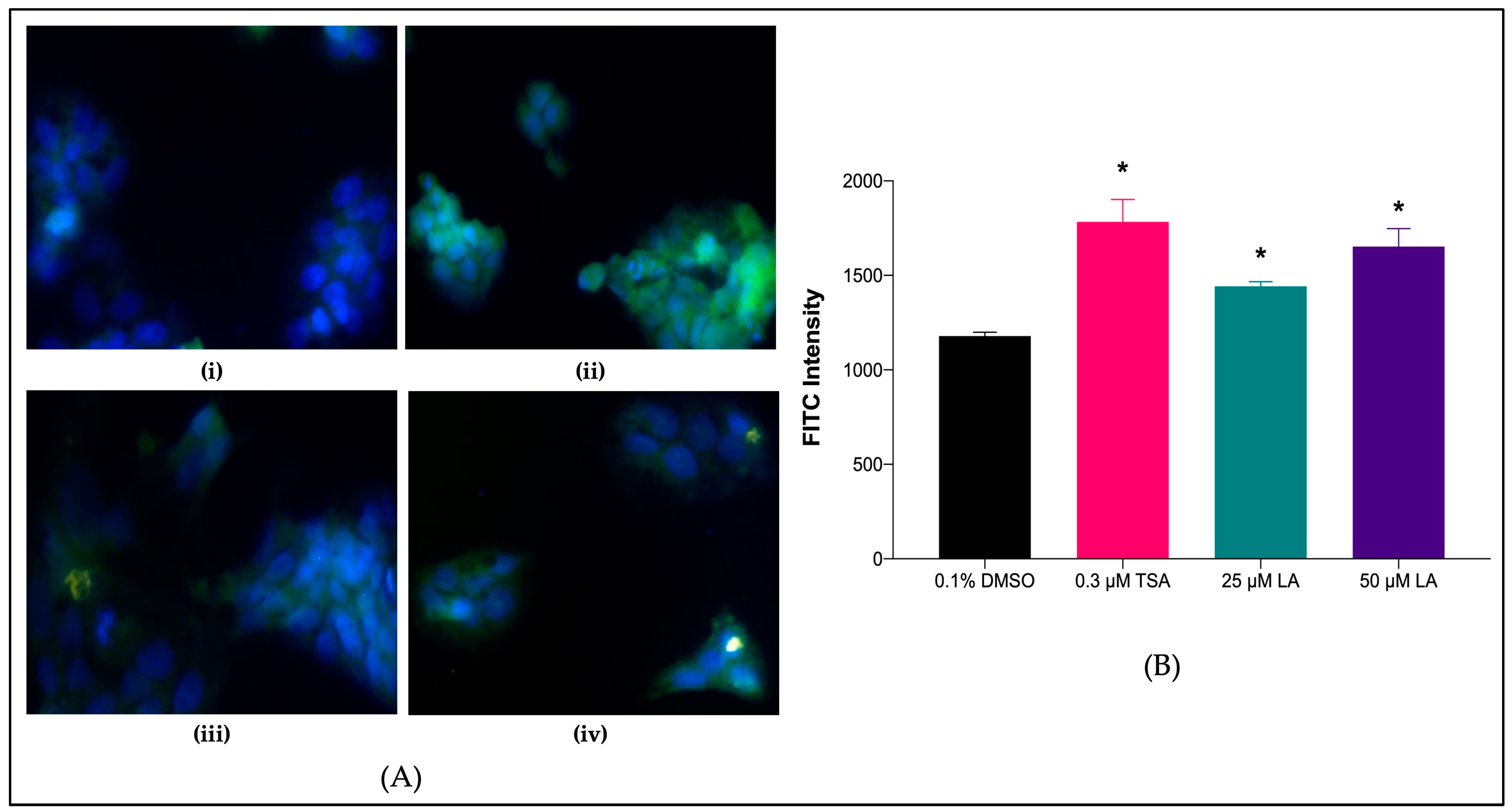
2.7.2. SR-BI Protein Levels in Treated HepG2 Cell Lines
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Preparation of the X. muta Extract
4.3. Preparation of X. muta Fractions
4.4. Cell Culture
4.5. Cytotoxicity Study Fraction by MTS Assay
4.6. SR-BI Transcriptional Activity of X. muta Fractions via Luciferase Assay
4.7. SR-BI Transcriptional Activity of Fraction-7 of X. muta via Luciferase Assay
4.8. Gas Chromatography-Mass Spectrometry (GC-MS) Analysis of Fraction-7 of X. muta
4.9. Induction of SR-BI Transcriptional Activity by Potential Fatty Acids
4.10. Treatment of HepG2 Cells with Fraction-7 of X. muta
4.10.1. Transfection of HepG2 Cells with SR-BI Promoter
4.10.2. Treatment of transfected HepG2 Cells with Fraction-7 of X. muta and LA
4.11. Extraction of Total RNA
4.11.1. RNA Extraction from HepG2 Cells
4.11.2. RNA Extraction from Tissue
4.11.3. Quantification of Total RNA
4.11.4. DNase Treatment of Total RNA
4.12. Rt-PCR of SR-BI Gene Expression
4.13. Determination of HDL-C Uptake and SR-BI Protein Cellular Localization
4.13.1. Determination of Lipid Uptake by Fluorometric Assay
4.13.2. Determination of SR-BI Protein Levels by Immunofluorescent Assay
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, X.; Zhao, X.D.; Bao, R.Q.; Yu, J.Y.; Zhang, G.X.; Chen, J.W. Effects of Extracts from Tiaozhi Granule and Its Components on Expression of Scavenger Receptor Class B Type i. Evid. Based Complement. Altern. Med. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, E.A. Regression of Atherosclerosis: The Journey from the Liver to the Plaque and Back. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegele, R.A.; Gidding, S.S.; Ginsberg, H.N.; McPherson, R.; Raal, F.J.; Rader, D.J.; Robinson, J.G.; Welty, F.K. Nonstatin Low-Density Lipoprotein-Lowering Therapy and Cardiovascular Risk Reduction-Statement from ATVB Council. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2269–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spence, J.D. Recent Advances in Pathogenesis, Assessment, and Treatment of Atherosclerosis. F1000Res 2016, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.G.; Stone, N.J. The 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Disease Risk: A New Paradigm Supported by More Evidence. Eur. Heart. J. 2015, 36, 2110–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd-Jones, D.M.; Goff, D.C.; Stone, N.J. Treatment of Blood Cholesterol to Reduce Risk for Atherosclerotic Cardiovascular Disease. Ann. Intern. Med. 2016, 164, 135–136. [Google Scholar] [CrossRef]
- Hou, J.R.; Wang, Y.H.; Zhong, Y.N.; Che, T.T.; Hu, Y.; Bao, J.; Meng, N. Protective Effect of Flavonoids from a Deep-Sea-Derived Arthrinium sp. against Ox-LDL-Induced Oxidative Injury through Activating the AKT/Nrf2/HO-1 Pathway in Vascular Endothelial Cells. Mar. Drugs 2021, 19, 712. [Google Scholar] [CrossRef]
- Jia, X.J.; Du, Y.; Jiang, H.J.; Li, Y.Z.; Xu, Y.N.; Si, S.Y.; Wang, L.; Hong, B. Identification of Novel Compounds Enhancing SR-BI MRNA Stability through High-Throughput Screening. SLAS Discov. 2020, 25, 397–408. [Google Scholar] [CrossRef]
- Zanoni, P.; Khetarpal, S.A.; Larach, D.B.; Hancock-Cerutti, W.F.; Millar, J.S.; Cuchel, M.; DerOhannessian, S.; Kontush, A.; Surendran, P.; Saleheen, D.; et al. Rare Variant in Scavenger Receptor BI Raises HDL Cholesterol and Increases Risk of Coronary Heart Disease. Science 2016, 351, 1166–1171. [Google Scholar] [CrossRef] [Green Version]
- Rosenson, R.S.; Brewer, H.B.; Ansell, B.J.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and Atherosclerotic Cardiovascular Disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef]
- Trajkovska, K.T.; Topuzovska, S. High-Density Lipoprotein Metabolism and Reverse Cholesterol Transport: Strategies for Raising HDL Cholesterol. Anatol. J. Cardiol. 2017, 18, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Sahebi, R.; Hassanian, S.M.; Ghayour-Mobarhan, M.; Farrokhi, E.; Rezayi, M.; Samadi, S.; Bahramian, S.; Ferns, G.A.; Avan, A. Scavenger Receptor Class B Type I as a Potential Risk Stratification Biomarker and Therapeutic Target in Cardiovascular Disease. J. Cell Physiol. 2019, 234, 16925–16932. [Google Scholar] [CrossRef] [PubMed]
- Andriani, Y.; Tengku-Muhammad, T.S.; Mohamad, H.; Saidin, J.; Syamsumir, D.F.; Chew, G.S.; Wahid, M.E.A. Phaleria Macrocarpa Boerl. (Thymelaeaceae) Leaves Increase SR-BI Expression and Reduce Cholesterol Levels in Rats Fed a High Cholesterol Diet. Molecules 2015, 20, 4410–4429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Kiem, P.; van Minh, C.; Nhiem, N.X.; Tai, B.H.; Yen, P.H.; Hang, D.T.T.; Dung, D.T.; Cuc, N.T.; Trang, D.T.; Yen, D.T.H.; et al. The Chemical Constituents and Biological Activity of Some Sponges in Northern Vietnam: A Review. Vietnam J. Chem. 2019, 57, 261–271. [Google Scholar] [CrossRef]
- Mehbub, M.F.; Lei, J.; Franco, C.; Zhang, W. Marine Sponge Derived Natural Products between 2001 and 2010: Trends and Opportunities for Discovery of Bioactives. Mar. Drugs 2014, 12, 4539–4577. [Google Scholar] [CrossRef] [Green Version]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2015, 32, 116–211. [Google Scholar] [CrossRef] [Green Version]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug Development from Marine Natural Products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Swierts, T.; Peijnenburg, K.T.C.A.; de Leeuw, C.; Cleary, D.F.R.; Hörnlein, C.; Setiawan, E.; Wörheide, G.; Erpenbeck, D.; de Voogd, N.J. Lock, Stock and Two Different Barrels: Comparing the Genetic Composition of Morphotypes of the Indo-Pacific Sponge Xestospongia Testudinaria. PLoS ONE 2013, 8, e74396. [Google Scholar] [CrossRef]
- Diaz, M.C.; Rützler, K. Sponges: An Essential Component of Caribbean Coral Reefs. Bull. Mar. Sci. 2001, 69, 535–546. [Google Scholar]
- Bell, J.J. The Functional Roles of Marine Sponges. Estuar. Coast Shelf Sci. 2008, 79, 341–353. [Google Scholar] [CrossRef]
- Ankisetty, S.; Slattery, M. Antibacterial Secondary Metabolites from the Cave Sponge Xestospongia sp. Mar. Drugs 2012, 10, 1037–1043. [Google Scholar] [CrossRef]
- Morinaka, B.I.; Skepper, C.K.; Molinski, T.F. Ene-Yne Tetrahydrofurans from the Sponge Xestospongia muta. Exploiting a Weak CD Effect for Assignment of Configuration. Org. Lett. 2007, 9, 1975–1978. [Google Scholar] [CrossRef]
- Kobayashi, M.; Chen, Y.J.; Aoki, S.; In, Y.; Ishida, T.; Kitagawa, I. Four New β-Carboline Alkaloids Isolated from Two Okinawan Marine Sponges of Xestospongia sp. and Haliclona sp. Tetrahedron 1995, 51, 3727–3736. [Google Scholar] [CrossRef]
- Agustina, S.; Karina, S.; Kurnianda, V.; Rahmi, R.; Khairunnisa, K.; Manzamine, C. An Alkaloid Indole as an Inhibitor of the Cancer Cells Adapted to Nutrient Starvation, from an Indonesian Marine Sponge of Xestospongia muta. IOP Conf. Ser. Earth Environ. Sci. 2018, 216, 1–6. [Google Scholar] [CrossRef]
- Harinantenaina Rakotondraibe, L.; Li, J.; Blasiak, L.; Hill, R.; Cassera, M. Antimalarial Dipeptide from a Streptomyces sp. Associate of the Sponge Xestospongia muta. Planta Med. 2014, 80, PB15. [Google Scholar] [CrossRef]
- Patil, A.D.; Kokke, W.C.; Cochran, S.; Francis, T.A.; Tomszek, T.; Westley, J.W. Brominated Polyacetylenic Acids from the Marine Sponge Xestospongia muta: Inhibitors of HIV Protease. J. Nat. Prod. 1992, 55, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Carballeira, N.M.; Maldonado, L. The Phospholipid Fatty Acids of the Marine Sponge Xestospongia muta. Lipids 1988, 23, 682–684. [Google Scholar] [CrossRef]
- Froyen, E.; Burns-Whitmore, B. The Effects of Linoleic Acid Consumption on Lipid Risk Markers for Cardiovascular Disease in Healthy Individuals: A Review of Human Intervention Trials. Nutrients 2020, 12, 2329. [Google Scholar] [CrossRef]
- van Schalkwijk, D.B.; Pasman, W.J.; Hendriks, H.F.J.; Verheij, E.R.; Rubingh, C.M.; van Bochove, K.; Vaes, W.H.J.; Adiels, M.; Freidig, A.P.; de Graaf, A.A. Dietary Medium Chain Fatty Acid Supplementation Leads to Reduced VLDL Lipolysis and Uptake Rates in Comparison to Linoleic Acid Supplementation. PLoS ONE 2014, 9, e100376. [Google Scholar] [CrossRef] [Green Version]
- Dias, C.B.; Amigo, N.; Wood, L.G.; Correig, X.; Garg, M.L. Effect of Diets Rich in Either Saturated Fat or N-6 Polyunsaturated Fatty Acids and Supplemented with Long-Chain n-3 Polyunsaturated Fatty Acids on Plasma Lipoprotein Profiles. Eur. J. Clin. Nutr. 2017, 71, 1297–1302. [Google Scholar] [CrossRef]
- Goyens, P.L.L.; Mensink, R.P. Human Nutrition and Metabolism the Dietary-Linolenic Acid to Linoleic Acid Ratio Does Not Affect the Serum Lipoprotein Profile in Human. J. Nutr. 2005, 135, 2799–2804. [Google Scholar] [CrossRef]
- French, M.A.; Sundram, K.; Clandinin, M.T. Cholesterolaemic Effect of Palmitic Acid in Relation to Other Dietary Fatty Acids. Asia Pac. J. Clin. Nutr. 2002, 11, S401–S407. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.H.; Tomasch, R.; Elmadfa, I. Impact of Diets Containing Corn Oil or Olive/Sunflower Oil Mixture on the Human Plasma and Lipoprotein Lipid Metabolism. Eur. J. Nutr. 2001, 40, 161–167. [Google Scholar] [CrossRef]
- Sanders, T.A.B.; Oakley, F.R.; Miller, G.J.; Mitropoulos, K.A.; Crook, D.; Oliver, M.F. Influence of N-6 versus n-3 Polyunsaturated Fatty Acids in Diets Low in Saturated Fatty Acids on Plasma Lipoproteins and Hemostatic Factors. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3449–3460. [Google Scholar] [CrossRef]
- Zock, P.L.; Katan, M.B. Hydrogenation Alternatives: Effects of Trans Fatty Acids and Stearic Acid versus Linoleic Acid on Serum Lipids and Lipoproteins in Humans. J. Lipid Res. 1992, 33, 399–410. [Google Scholar] [CrossRef]
- Azemi, N.A.; Ismail, N.; Abu-Bakar, L.; Muhammad, T.-S.T. Sterol Composition And Anti-Atherosclerosis Effects of Xestospongia muta Extracts By Increasing Transcriptional Activity of Sr-B1 Promoter. Int. J. Pharm. Sci. Res. 2019, 10, 2892–2897. [Google Scholar] [CrossRef]
- Dung, D.T.; Hang, D.T.T.; Yen, P.H.; Quang, T.H.; Nhiem, N.X.; Tai, B.H.; Minh, C.V.; Kim, Y.C.; Kim, D.C.; Oh, H.; et al. Macrocyclic Bis-Quinolizidine Alkaloids from Xestospongia muta. Nat. Prod. Res. 2019, 33, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Rubio, I.; Martín, C.; Civeira, F.; Cenarro, A. Sr-B1, a Key Receptor Involved in the Progression of Cardiovascular Disease: A Perspective from Mice and Human Genetic Studies. Biomedicines 2021, 9, 612. [Google Scholar] [CrossRef] [PubMed]
- Rothblat, G.H.; de La Llera-Moya, M.; Atger, V.; Kellner-Weibel, G.; Williams, D.L.; Phillips, M.C. Cell Cholesterol Efflux: Integration of Old and New Observations Provides New Insights. J. Lipid. Res. 1999, 40, 781–796. [Google Scholar] [CrossRef]
- Ueda, Y.; Royer, L.; Gong, E.; Zhang, J.; Cooper, P.N.; Francone, O.; Rubin, E.M. Lower Plasma Levels and Accelerated Clearance of High Density Lipoprotein (HDL) and Non-HDL Cholesterol in Scavenger Receptor Class B Type I Transgenic Mice. J. Biol. Chem. 1999, 274, 7165–7171. [Google Scholar] [CrossRef] [Green Version]
- Kozarsky, K.F.; Donahee, M.H.; Rigotti, A.; Iqbal, S.N.; Edelman, E.R.; Krieger, M. Overexpression of the HDL Receptor SR-BI Alters Plasma HDL and Bile Cholesterol Levels. Nature 1997, 387, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Farvid, M.S.; Ding, M.; Pan, A.; Sun, Q.; Chiuve, S.E.; Steffen, L.M.; Willett, W.C.; Hu, F.B. Dietary Linoleic Acid and Risk of Coronary Heart Disease: A Systematic Review and Meta-Analysis of Prospective Cohort Studies. Circulation 2014, 130, 1568–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azemi, N.-A.; Abu-Bakar, L.; Ismail, N.; Sevakumaran, V.; Tengku-Muhammad, T.-S. Linoleic Acid Treatment Increases the Expression of Scavenger Receptor Class B Type 1 (SR-B1) in in-Vitro Model. Atherosclerosis 2021, 331, e128. [Google Scholar] [CrossRef]
- Garelnabi, M.; Ainsworth, G.; Mahini, H.; Jamil, N.; Ochin, C. Dietary Oxidized Linoleic Acid Modulates Plasma Lipids beyond Triglycerides Metabolism. J. Lipids 2017, 2017, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, R.L.; Huang, C.X.; Bao, J.L.; Jiang, J.Y.; Zhang, B.; Zhou, S.X.; Cai, W.B.; Wang, H.; Wang, J.F.; Zhang, Y.L. CC-Chemokine Ligand 2 (CCL2) Suppresses High Density Lipoprotein (HDL) Internalization and Cholesterol Efflux via CC-Chemokine Receptor 2 (CCR2) Induction and P42/44 Mitogen-Activated Protein Kinase (MAPK) Activation in Human Endothelial Cells. J. Biol. Chem. 2016, 291, 19532–19544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.J.; Hu, J.; Hu, Z.; Kraemer, F.B.; Azhar, S. Scavenger Receptor Class B Type i (SR-BI): A Versatile Receptor with Multiple Functions and Actions. Metabolism 2014, 63, 875–886. [Google Scholar] [CrossRef]
- Iguchi, K.; Shimura, H.; Taira, S.; Yokoo, C.; Matsumoto, K.; Yamada, Y. Aragusterol B and D, New 26,27-Cyclosterols from the Okinawan Marine Sponge of the Genus Xestospongia. J. Org. Chem. 1994, 59, 7499–7502. [Google Scholar] [CrossRef]
- Ansari, K.I.; Kasiri, S.; Hussain, I.; Morris Bobzean, S.A.; Perrotti, L.I.; Mandal, S.S. MLL Histone Methylases Regulate Expression of HDLR-SR-B1 in Presence of Estrogen and Control Plasma Cholesterol in Vivo. Mol. Endocrinol. 2013, 27, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Noh, H.; Eun, Y.O.; Ji, Y.S.; Mi, R.Y.; Young, O.K.; Ha, H.; Hi, B.L. Histone Deacetylase-2 Is a Key Regulator of Diabetes- and Transforming Growth Factor-Β1-Induced Renal Injury. Am. J. Physiol. Renal Physiol. 2009, 297, F729–F739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.; Zhang, X.; Jia, K.; Zhang, C.; Zhu, L.; Cheng, M.; Li, F.; Zhao, S.; Hao, J. Trichostatin A Increases BDNF Protein Expression by Improving XBP-1s/ATF6/GRP78 Axis in Schwann Cells of Diabetic Peripheral Neuropathy. Biomed. Pharmacother. 2021, 133, 111062. [Google Scholar] [CrossRef]
- Ling, T.; Xie, J.; Shen, Y.S.; Qiao, M.; Yang, H.; Sun, D.Y.; Qian, K.J. Trichostatin A Exerts Anti-Inflammation Functions in LPS-Induced Acute Lung Injury Model through Inhibiting TNF-α and Upregulating MicorRNA-146a Expression. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3935–3942. [Google Scholar] [CrossRef]
- Choi, J.H.; Oh, S.W.; Kang, M.S.; Kwon, H.J.; Oh, G.T.; Kim, D.Y. Trichostatin A Attenuates Airway Inflammation in Mouse Asthma Model. Clin. Exp. Allergy 2005, 35, 89–96. [Google Scholar] [CrossRef]
- Nunes, M.J.; Moutinho, M.; Gama, M.J.; Rodrigues, C.M.P.; Rodrigues, E. Histone Deacetylase Inhibition Decreases Cholesterol Levels in Neuronal Cells by Modulating Key Genes in Cholesterol Synthesis, Uptake and Efflux. PLoS ONE 2013, 8, e53394. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.G.; Cho, G.W. Trichostatin a Modulates Intracellular Reactive Oxygen Species through SOD2 and FOXO1 in Human Bone Marrow-Mesenchymal Stem Cells. Cell Biochem. Funct. 2015, 33, 37–43. [Google Scholar] [CrossRef]
- Guo, Y.; Li, Z.; Shi, C.; Li, J.; Yao, M.; Chen, X. Trichostatin A Attenuates Oxidative Stress-Mediated Myocardial Injury through the FoxO3a Signaling Pathway. Int. J. Mol. Med. 2017, 40, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Bouyahya, A.; el Omari, N.; Bakha, M.; Aanniz, T.; el Menyiy, N.; el Hachlafi, N.; el Baaboua, A.; El-Shazly, M.; Alshahrani, M.M.; al Awadh, A.A.; et al. Pharmacological Properties of Trichostatin A, Focusing on the Anticancer Potential: A Comprehensive Review. Pharmaceuticals 2022, 15, 1235. [Google Scholar] [CrossRef]
- Chittur, S.V.; Sangster-Guity, N.; McCormick, P.J. Histone Deacetylase Inhibitors: A New Mode for Inhibition of Cholesterol Metabolism. BMC Genomics 2008, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.W.; Hong, B.H.; Bai, K.K.; Tan, R.; Yang, T.; Sun, J.P.; Yi, R.Z.; Wu, H. Cis- and Trans-Palmitoleic Acid Isomers Regulate Cholesterol Metabolism in Different Ways. Front. Pharmacol. 2020, 11, 602115. [Google Scholar] [CrossRef]
- Matthan, N.R.; Dillard, A.; Lecker, J.L.; Ip, B.; Lichtenstein, A.H. Effects of Dietary Palmitoleic Acid on Plasma Lipoprotein Profile and Aortic Cholesterol Accumulation Are Similar to Those of Other Unsaturated Fatty Acids in the F1B Golden Syrian Hamster. J. Nutr. 2009, 139, 215–221. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fractions of X. muta | Concentration of Fractions at 50% Cell Viability (μg/mL) | SR-BI Fold Change |
|---|---|---|
| Fraction 1 | >100 | 0.62 ± 0.05 |
| Fraction 2 | 55.30 ± 3.76 | 0.56 ± 0.02 |
| Fraction 3 | 15.63 ± 1.66 | - |
| Fraction 4 | 20.48 ± 1.65 | - |
| Fraction 5 | >100 | 0.51 ± 0.25 |
| Fraction 6 | >100 | 1.21 ± 0.36 |
| Fraction 7 | 82.27 ± 1.26 | 1.64 ± 0.01 |
| Gene | Primers | Sequence |
|---|---|---|
| SR-BI | Forward | 5′-GATGATGGTCCCGATAGAGG-3′ |
| Luciferase | Reverse | 5′-GGTAGCTTCTTTTGCACGTTG-3′ |
| Gene | Primers | Sequence |
|---|---|---|
| hSR-BI | Forward | 5′-CTG TGG GTG AGA TCA TGT GG-3′ |
| Reverse | 5′-GTT CCA CTT GTC CAC GAG GT-3′ | |
| h-actin | Forward | 5′-TCA CCC TGA AGT ACC CCA TC-3′ |
| Reverse | 5′-CCA TCT CTT GCT CGA AGT CC-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azemi, N.A.; Azemi, A.K.; Abu-Bakar, L.; Sevakumaran, V.; Muhammad, T.S.T.; Ismail, N. Xestospongia muta Fraction-7 and Linoleic Acid: Effects on SR-BI Gene Expression and HDL Cholesterol Uptake. Mar. Drugs 2022, 20, 762. https://doi.org/10.3390/md20120762
Azemi NA, Azemi AK, Abu-Bakar L, Sevakumaran V, Muhammad TST, Ismail N. Xestospongia muta Fraction-7 and Linoleic Acid: Effects on SR-BI Gene Expression and HDL Cholesterol Uptake. Marine Drugs. 2022; 20(12):762. https://doi.org/10.3390/md20120762
Chicago/Turabian StyleAzemi, Nurul Adila, Ahmad Khusairi Azemi, Luqman Abu-Bakar, Vigneswari Sevakumaran, Tengku Sifzizul Tengku Muhammad, and Noraznawati Ismail. 2022. "Xestospongia muta Fraction-7 and Linoleic Acid: Effects on SR-BI Gene Expression and HDL Cholesterol Uptake" Marine Drugs 20, no. 12: 762. https://doi.org/10.3390/md20120762