
Marine-Derived Compounds Targeting Topoisomerase II in Cancer Cells: A Review
 ,
,  , ,
, , 
Abstract
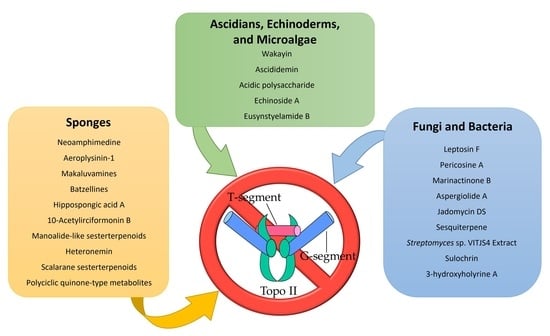
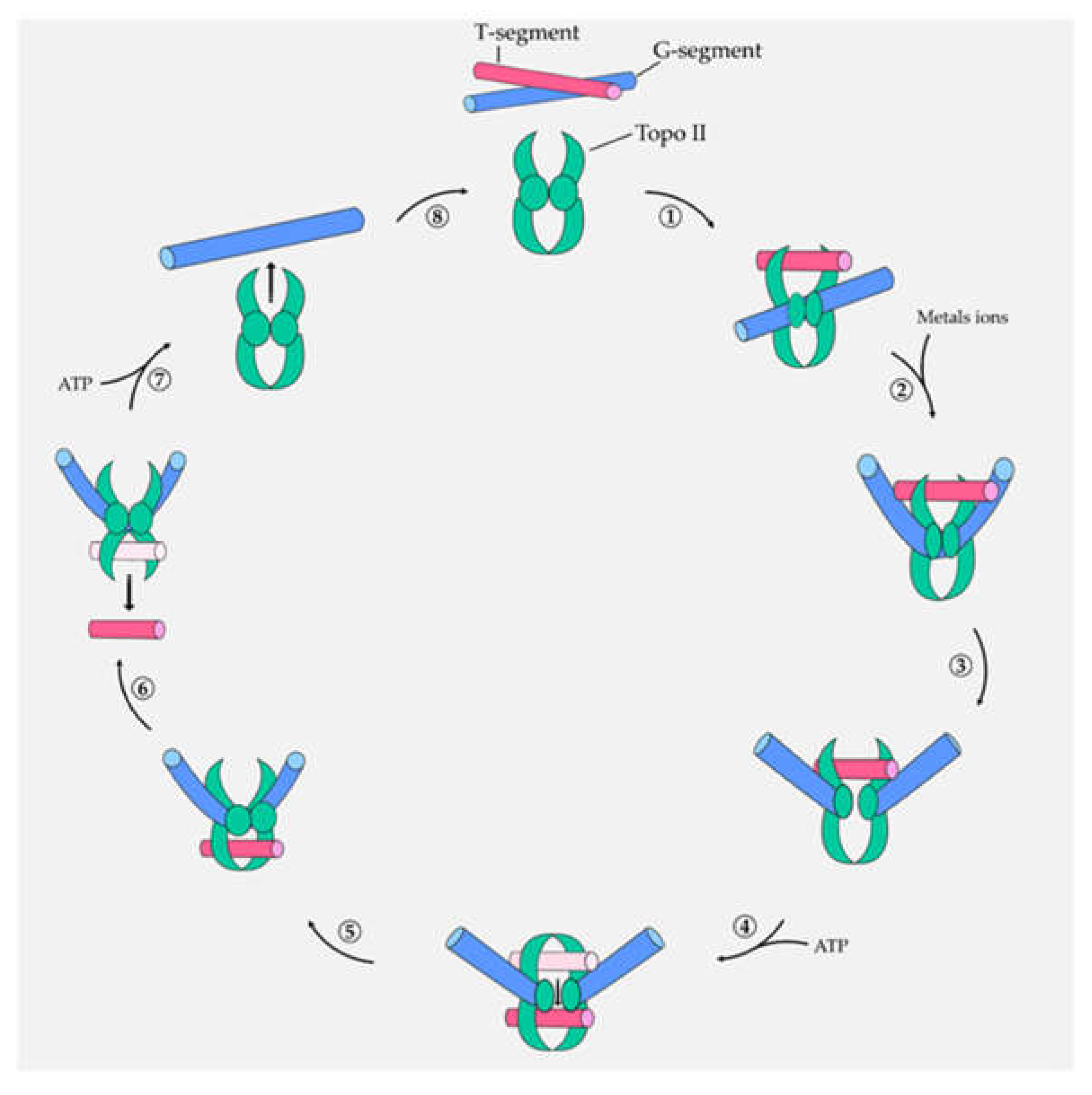
:
1. Introduction
2. Topo II Inhibitors from Marine Sponges
2.1. Neoamphimedine and Amphimedine
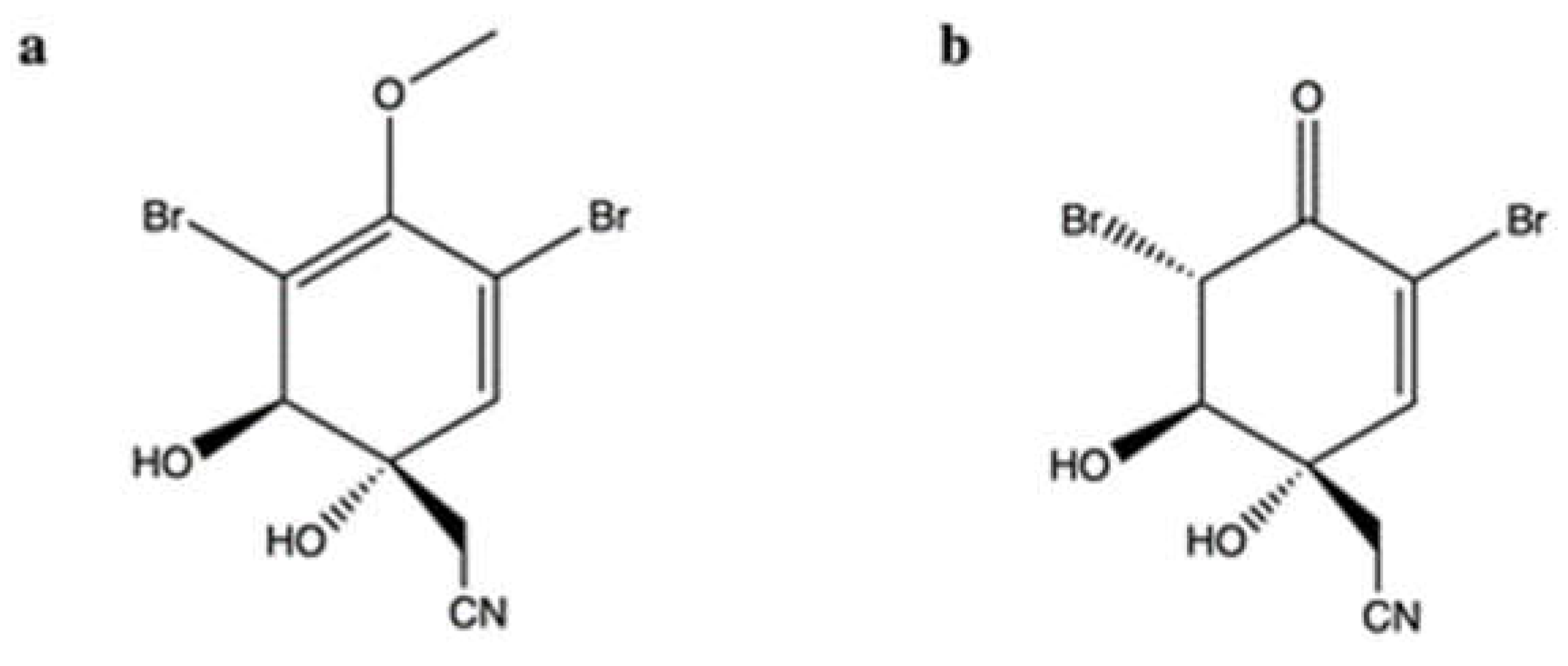
2.2. Aeroplysinin-1 and Its Oxidized Derivative
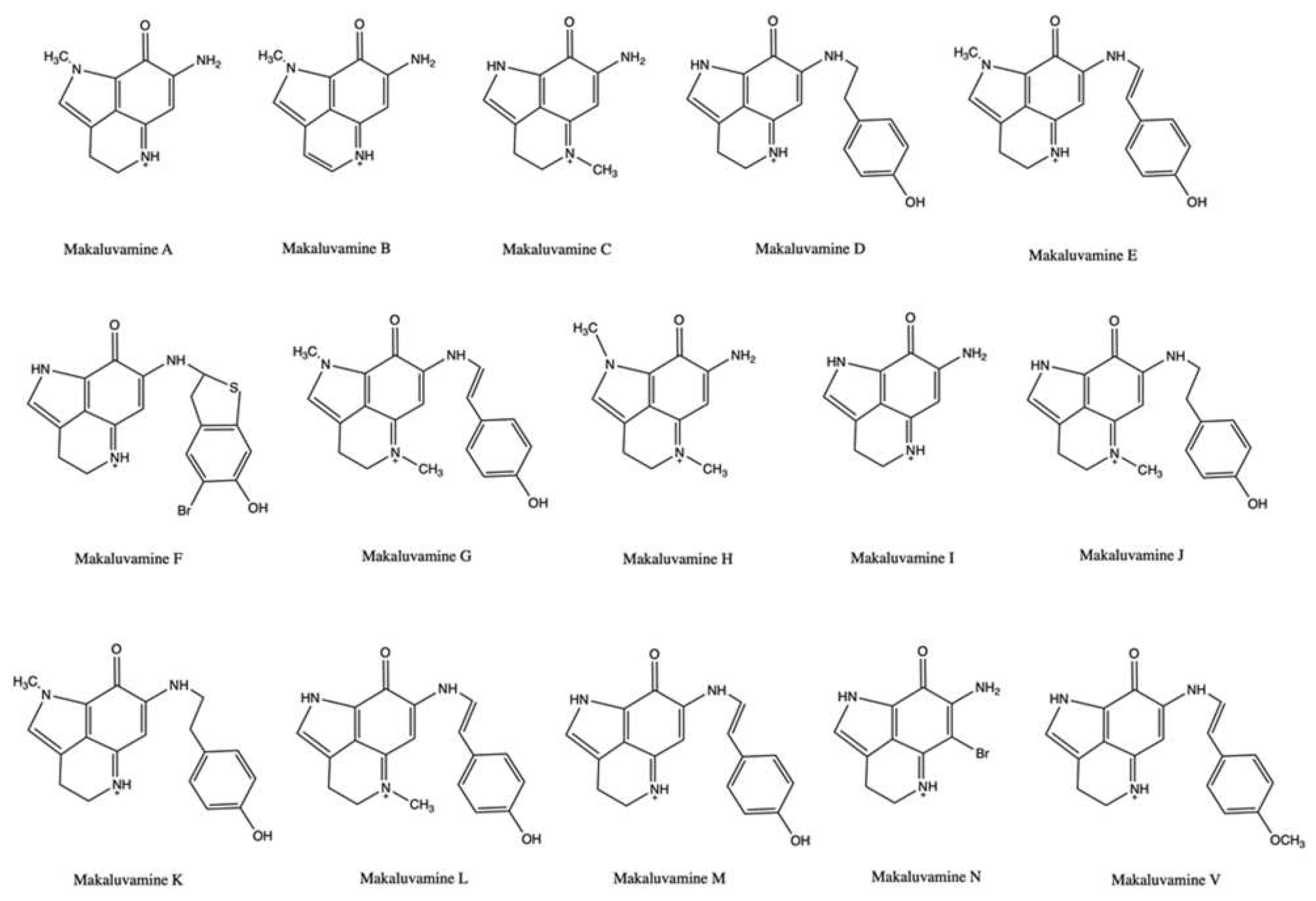
2.3. Makaluvamines
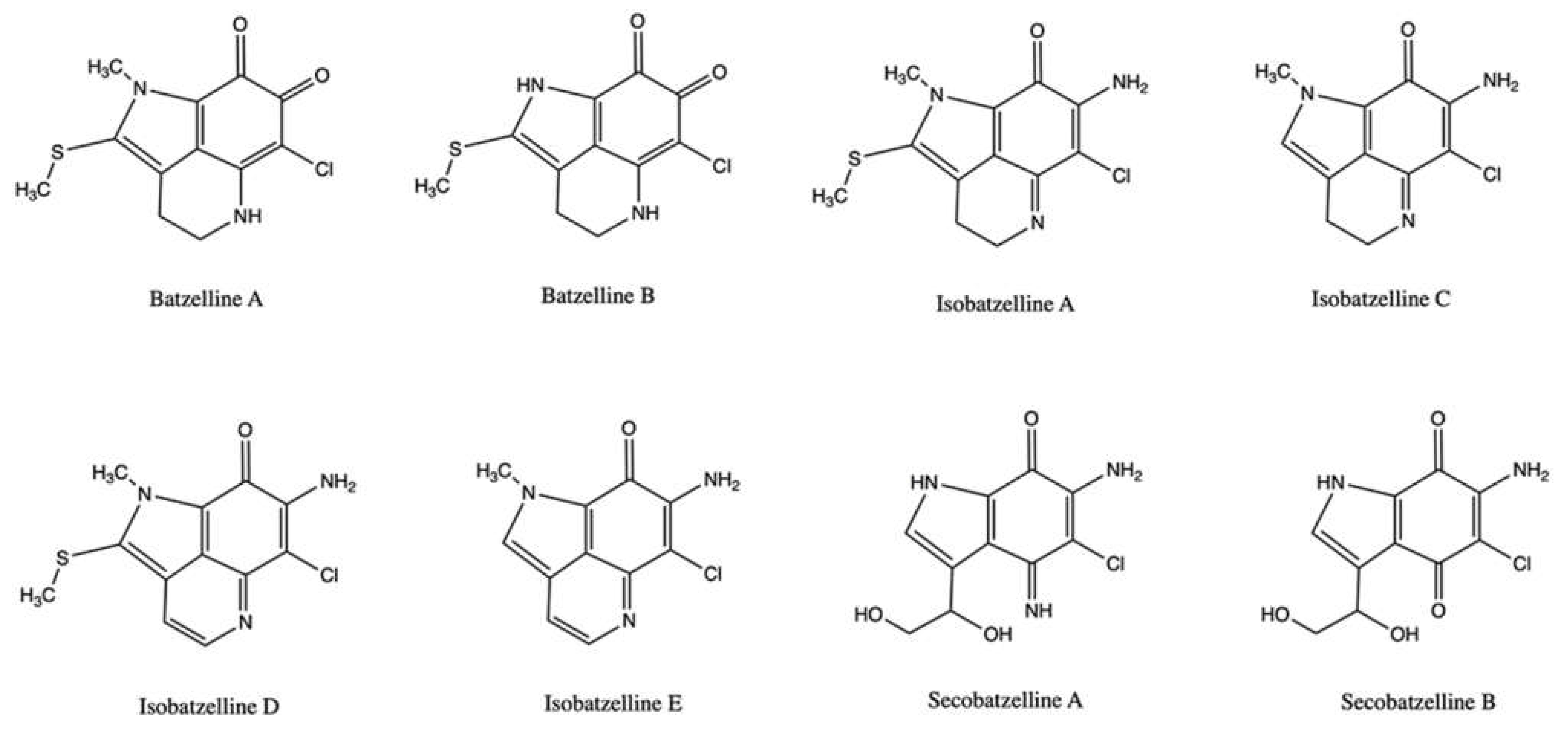
2.4. Batzellines
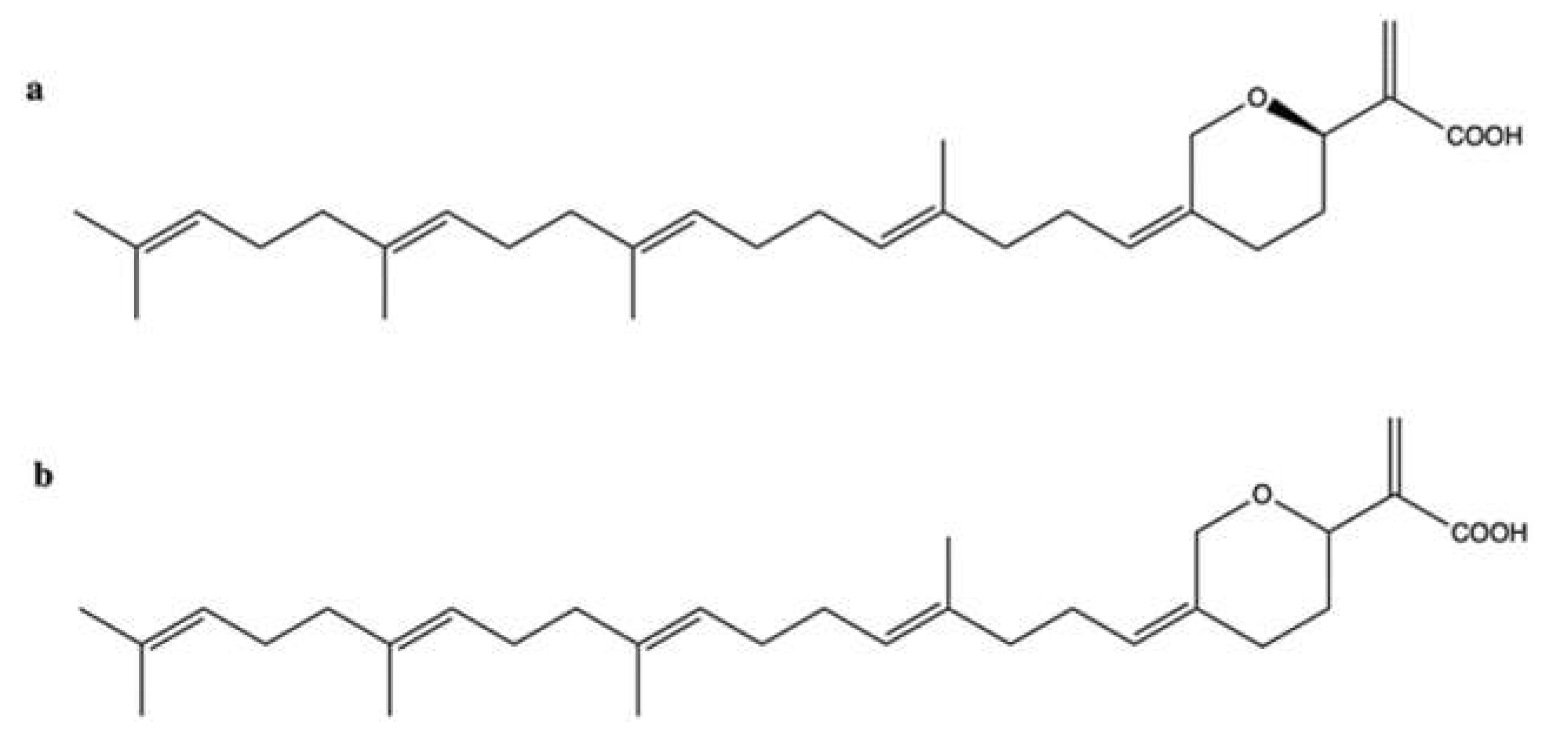
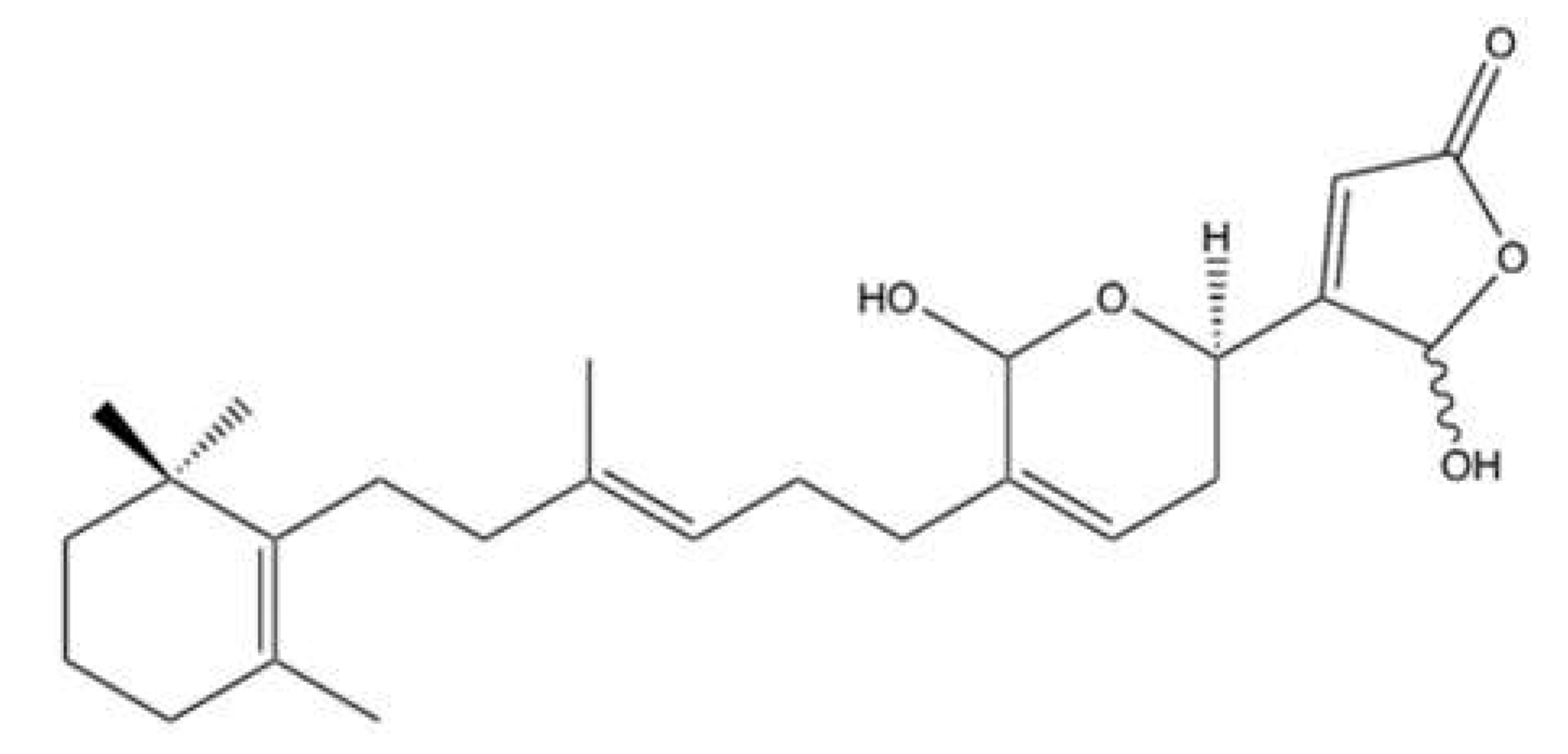
2.5. Hippospongic Acid A
2.6. 10-Acetylirciformonin B
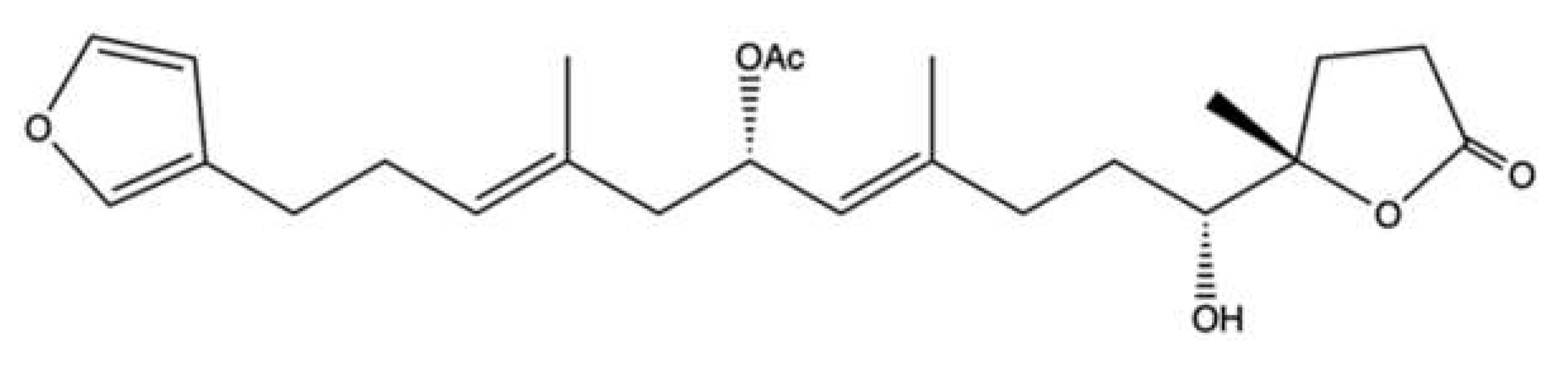
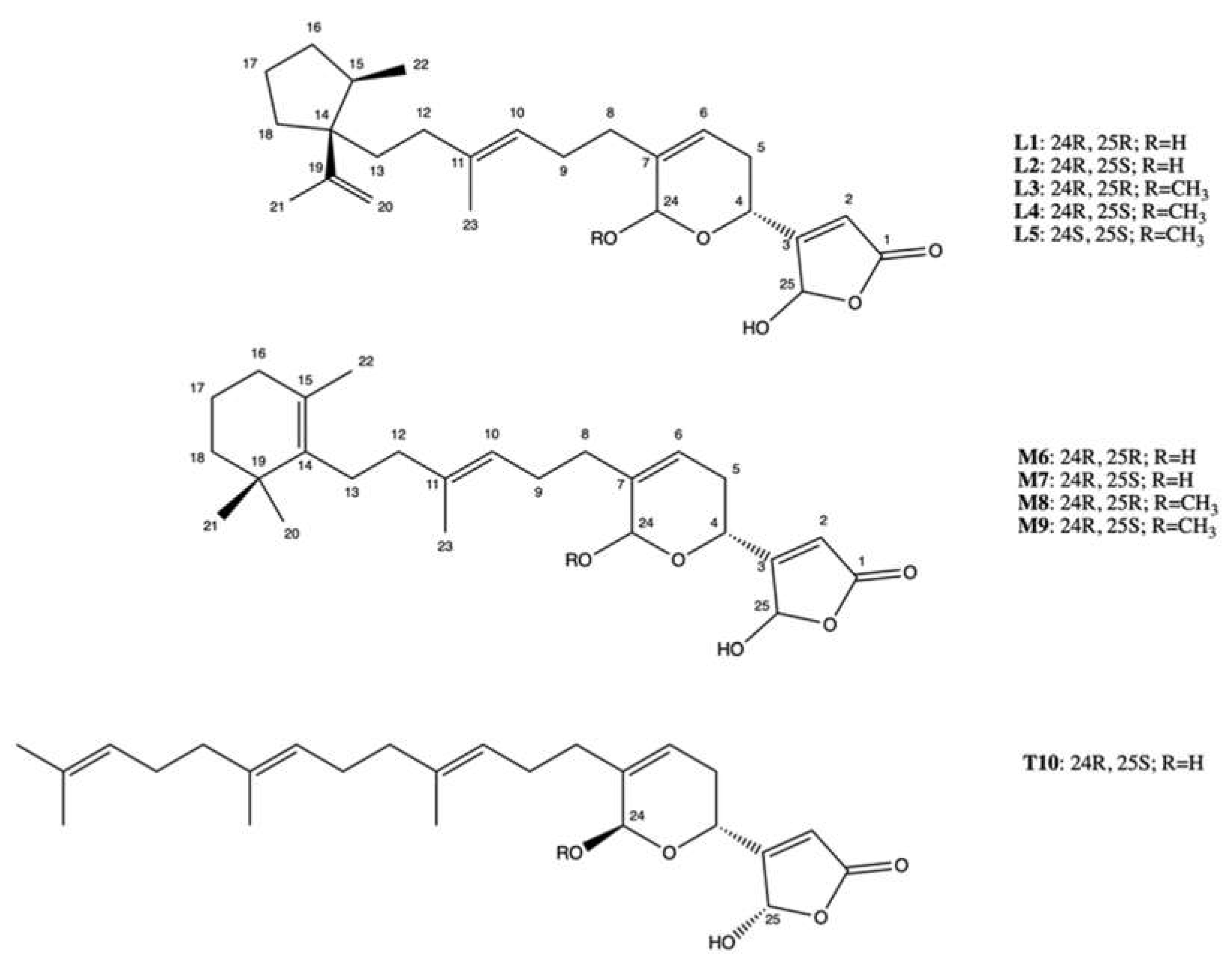
2.7. Manoalide-Like Sesterterpenoids
2.8. Heteronemin
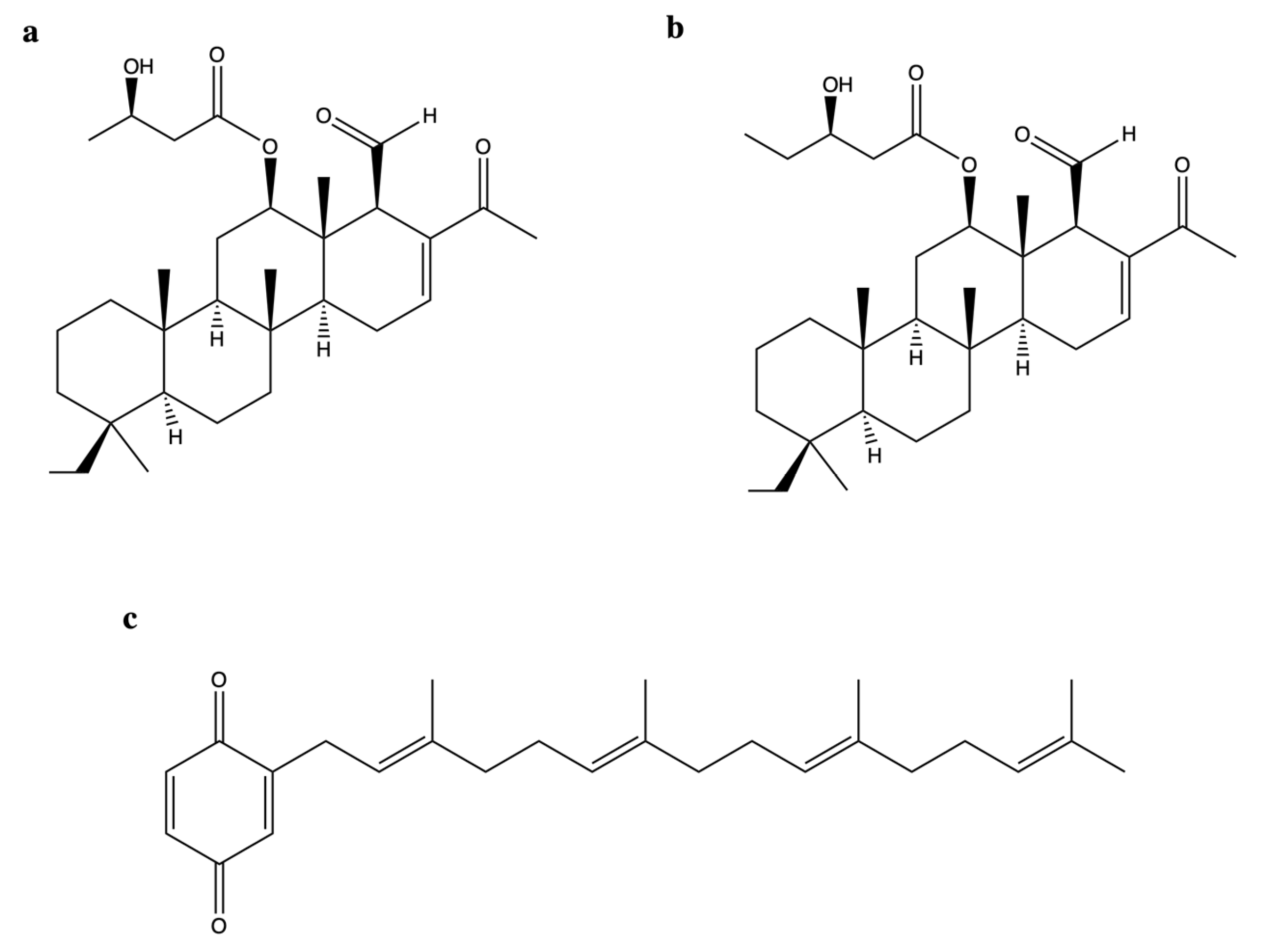
2.9. Scalarane Sesterterpenoids
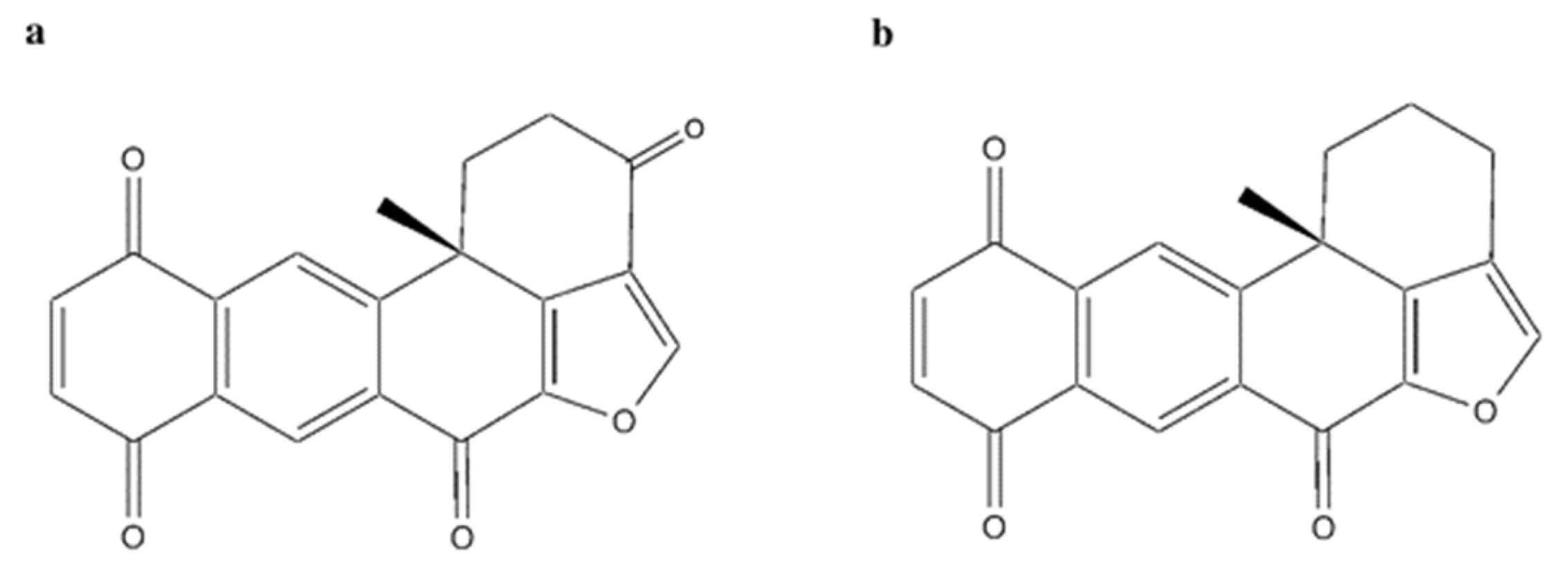
2.10. Polycyclic Quinone-Type Metabolites

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Sponge Species (Sp.) | Topo II Inhibitory Activity | Antitumor Effect(s) | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Assay | IC50/IC90 a or Range of Concentrations Tested | Outcomes | Experimental Model | Cytotoxic Activity (IC50 b) | Other Antitumor Mechanism(s) | |||
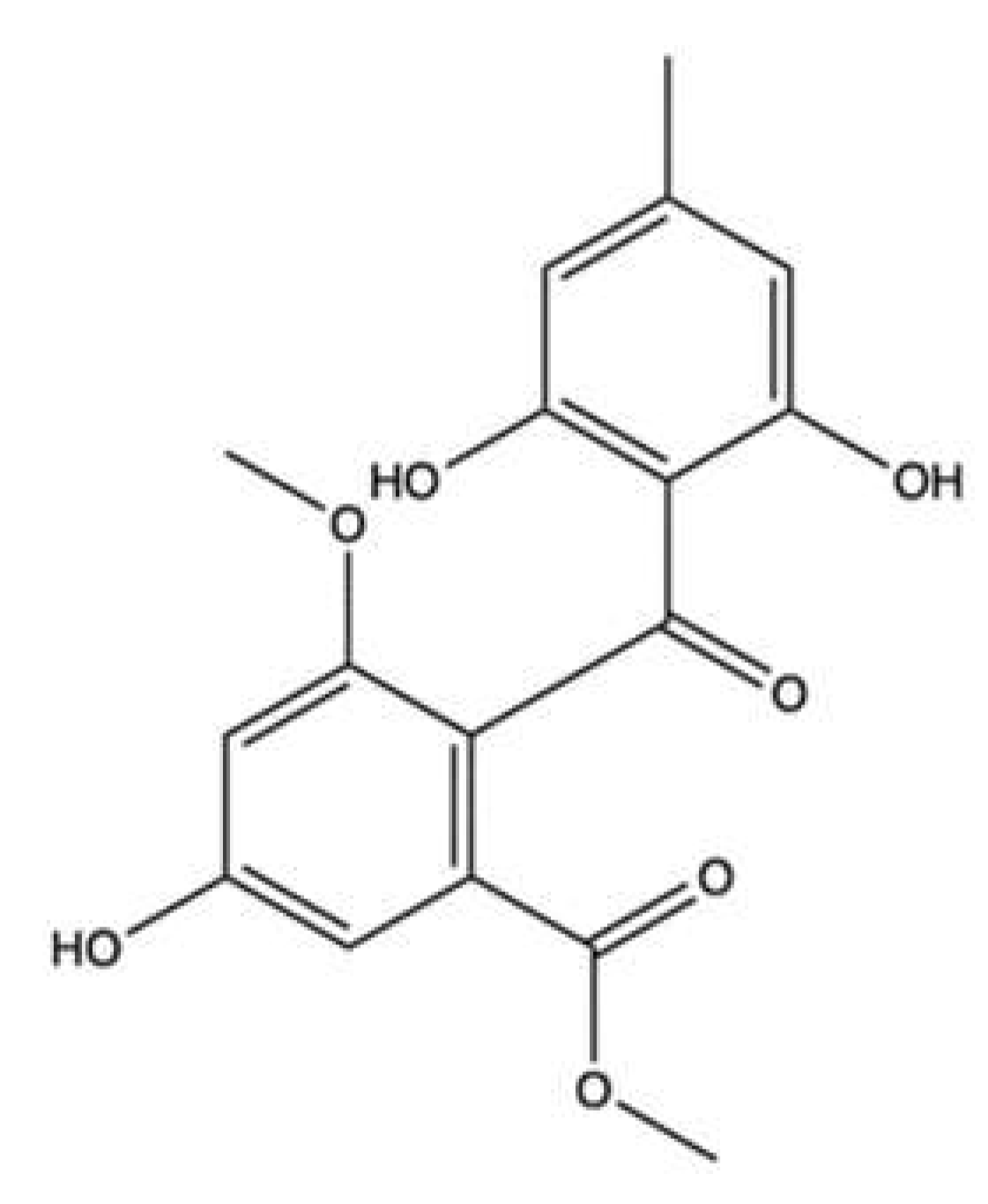
| (1′R,5′S,6′S)-2-(3′,5′-dibromo-1′,6′-dihydroxy-4′-oxocyclohex-2′-enyl) acetonitrile | Pseudoceratina sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA and human topo II | 2.5–10 μg/mL | ↓ DNA relaxation in presence of topo IIα | K562 | IC50 (72 h): 1.4 μg/mL | Apoptosis (↑ cleaved PARP, ↑ cleaved caspase-3, ↑ XIAP protein expression) DNA damage (↑ p-ATM, ↑ p-ATR, ↑ γH2A.X protein expression, ↑ p-BRCA, ↑ p-Chk2 protein expression) Oxidative stress (↑ ROS) ↓ IKK/NFκB pathway ↑ PI3K/Akt pathway | [35] |
| HeLa | IC50 (72 h): 4.8 μg/mL | |||||||
| MCF-7 | IC50 (72 h):1.9 μg/mL | |||||||
| MDA-MB-231 | IC50 (72 h): 5.5 μg/mL | |||||||
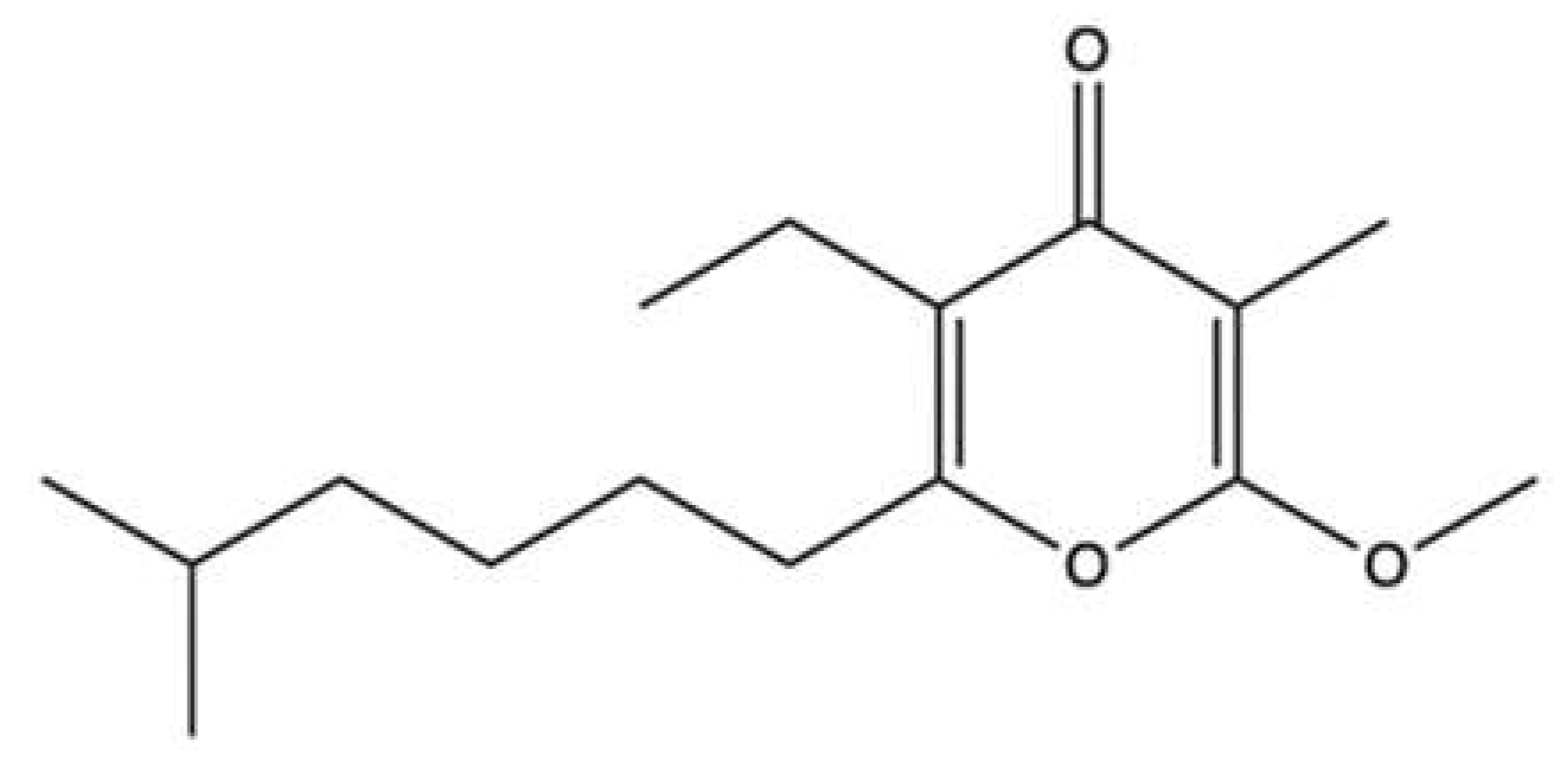
| 2-tetraprenil-1,4-benzoquinone | Carteriospongia sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA and human topo II | IC50: 0.43 μg/mL | ↓ DNA relaxation and formation of supercoiled DNA products in presence of topo IIα | Molt-4 | IC50 (72 h): 0.34 μg/mL | Apoptosis (↓ MMP) | [74] |
| K562 | IC50 b (72 h): 0.70 μg/mL | |||||||
| HL-60 | IC50 (72 h): 0.42 μg/mL | |||||||
| U937 | IC50 (72 h): 0.65 μg/mL | |||||||
| Sup-T1 | IC50 (72 h): 0.33 μg/mL | |||||||
| Ca9-22 | IC50 (72 h): 0.97 μg/mL | |||||||
| Cal-27 | IC50 (72 h): 0.51 μg/mL | |||||||
| LNCaP | IC50 (72 h): >20 μg/mL | |||||||
| DLD-1 | IC50 (72 h): 15.41 μg/mL | |||||||
| T-47D | IC50 (72 h): 1.06 μg/mL | |||||||
| Immunodeficient athymic mice bearing Molt-4 xenograft | ↓ Tumor growth (1.14 μg/g/day, for 33 days) | |||||||
| 10-acetylirciformonin B | Ircinia sp. | Cell-free DNA cleavage assay using an enzyme-mediated supercoiled pHOT1 plasmid DNA and human topo II | 1.5–6.0 μM | ↓ DNA relaxation and formation of supercoiled DNA products in the presence of topo IIα | HL-60 | Not indicated | Apoptosis (↓ MMP; ↓ Bcl-2, ↓ Bcl-xL, ↓ XIAP, ↓ survinin, ↑ BAX, ↑ cleaved PARP protein expression, ↑ cyt c release) ↓ p-Akt, ↓ p-PTEN, ↓ Src, ↓ HKII, ↓ PKM 2, ↑ p-ERK, ↑ p-38, ↑ p-JNK, ↑ p-GSK 3β protein expression Oxidative stress (↑ ROS) | [57] |
| Western Blotting on HL-60 cells | 3.0 μM | ↓ Topo IIα protein expression | ||||||
| 12β-(3′β-hydroxybutanoyloxy)-20,24-dimethyl-24-oxo-scalara- 16-en-25-al | Carteriospongia sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA and human topo II | IC50: 1.98 μg/mL | ↓ DNA relaxation and formation of supercoiled DNA products in presence of topo IIα | K562 | IC50 (72 h): 0.01 μg/mL | [74] | |
| Molt-4 | IC50 (72 h): 0.01 μg/mL | Apoptosis (↓ MMP; ↑ cleaved caspase-8, ↑ cleaved caspase-9, ↑ cleaved PARP protein expression) ↑ DNA damage (↑ γH2AX protein expression, ↑ DNA DSBs) Oxidative stress (↑ ROS) ↑ ER stress (↑ Ca2+ release; ↑ IRE 1α, ↑ Bip, ↑ CHOP, ↑ Grp94, ↑ Hsp70, ↑ ATF6, ↓ PERK protein expression) ↓ Hsp90 (↓ Akt, ↓ p70S6k, ↓ NFκB, ↓ Raf-1, ↓ p-GSK3β, ↓ XIAP, ↓ MDM 2 ↓ Rb2, ↓ CDK4 ↓Cyclin D3, ↓ HIF 1 ↓ HSF1; ↑ Hsp70 protein expression) | ||||||
| HL-60 | IC50 (72 h): 0.01 μg/mL | |||||||
| U937 | IC50 (72 h): 0.01 μg/mL | |||||||
| Sup-T1 | IC50 (72 h): 0.13 μg/mL | |||||||
| Ca9-22 | IC50 (72 h): 0.10 μg/mL | |||||||
| Cal-27 | IC50 (72 h): 0.56 μg/mL | |||||||
| LNCaP | IC50 (72 h): 13.87 μg/mL | |||||||
| DLD-1 | IC50 (72 h): 2.33 μg/mL | |||||||
| T-47D | IC50 (72 h): 2.19 μg/mL | |||||||
| 12β-(3′β-hydroxypentanoyloxy)-20,24-dimethyl-24-oxo-scalara-16-en-25-al | Carteriospongia sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA and human topo II | IC50: 0.37 μg/mL | ↓ DNA relaxation | K562 | IC50 (72 h): 0.35 μg/mL | [74] | |
| Molt-4 | IC50 (72 h): 0.30 μg/mL | Apoptosis (↓ MMP) DNA damage (↑ γH2AX protein expression) | ||||||
| HL-60 | IC50 (72 h): 0.22 μg/mL | |||||||
| U937 | IC50 (72 h): 0.61 μg/mL | |||||||
| Sup-T1 | IC50 (72 h): 0.42 μg/mL | |||||||
| Ca9-22 | IC50 (72 h): 1.48 μg/mL | |||||||
| Cal-27 | IC50 (72 h): 3.17 μg/mL | |||||||
| LNCaP | IC50 (72 h): >20 μg/mL | |||||||
| DLD-1 | IC50 (72 h): 1.71 μg/mL | |||||||
| T-47D | IC50 (72 h): 1.87 μg/mL | |||||||
| 14-methoxy-xestoquinone | Xestopongia sp. | Cell-free DNA relaxation assay with supercoiled pBR322 DNA plasmid and topo II of drosophila | IC90: 143 μM | ↓ DNA relaxation | HCT-116 | IC50 (18 + 72 h): 28 μM | [77] | |
| CHO xrs-6 c | IC50 (18 + 72 h): 4.3 μM | |||||||
| 14-chloro-15-hydroxyxestoquinone | Xestopongia sp. | Cell-free decatenation reaction of kinetoplast DNA | IC90: 110 μM | ↓ DNA decatenation | HCT-116 | IC50 (18 + 72 h): 33 μM | [77] | |
| Cell-free DNA relaxation assay with supercoiled pBR322 DNA and topo II of drosophila | IC90: 135 μM | ↓ DNA relaxation | CHO xrs-6 c | IC50 (18 + 72 h): 27 μM | ||||
| 15-methoxy-xestoquinone | Xestopongia sp. | Cell-free DNA relaxation assay with supercoiled pBR322 DNA and Topo II of drosophila | IC90: 143 μM | ↓ DNA relaxation | HCT-116 | IC50 (18 + 72 h): 28 μM | [77] | |
| CHO xrs-6 c | IC50 (18 + 72 h): 4.3 μM | |||||||
| 15-chloro-14-hydroxyxestoquinone | Xestopongia sp. | Cell-free decatenation reaction of kinetoplast DNA | IC90: 110 μM | ↓ DNA decatenation | HCT-116 | IC50 (18 + 72 h): 33 μM | [77] | |
| Cell-free DNA relaxation assay with supercoiled pBR322 DNA and topo II of drosophila | IC90: 135 μM | ↓ DNA relaxation | CHO xrs-6 c | IC50 (18 + 72 h): 27 μM | ||||
| 24R, 25S-Manoalide | Luffariella sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA and human topo II | IC50: 1.18 μM | ↓ DNA relaxation | Molt-4 | IC50 (72 h): 0.82 μM | Apoptosis (↓ MMP; ↑ cleaved PARP, ↑ cleaved caspase-3, ↑ cleaved caspase-8, ↑ cleaved caspase-9 protein expression) DNA damage (↑ p-ATM, ↑ p-Chk2, ↑ γH2AX protein expression, ↑ DSBs) Oxidative stress (↑ ROS) | [61] |
| K562 | IC50 (72 h): 7.67 μM | |||||||
| Sup-T1 | IC50 (72 h): 1.35 μM | |||||||
| U937 | IC50 (72 h): 1.56 μM | |||||||
| Adociaquinone A | Xestopongia sp. | Cell-free DNA relaxation assay with supercoiled pBR322 DNA and topo II of drosophila | IC90: 118 μM | ↓ DNA relaxation | HCT-116 | IC50 (18 + 72 h): 24 μM | [77] | |
| CHO xrs-6 c | IC50 (18 + 72 h): 78 μM | |||||||
| Adociaquinone B | Xestopongia sp. | Cell-free decatenation reaction of kinetoplast DNA | IC90: <11 μM | ↓ DNA decatenation | HCT-116 | IC50 (18 + 72 h): 21 μM | [77] | |
| Cell-free DNA relaxation assay with supercoiled pBR322 DNA and topo II of drosophila | IC90: 78 μM | ↓ DNA relaxation | CHO xrs-6 c | IC50 (18 + 72 h): 23 μM | ||||
| KSDS assay | / | Formation of enzyme-DNA cleavable complex | ||||||
| Aeroplysinin 1 | Pseudoceratina sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA and human topo II | IC50: 1.37 μM | ↓ DNA relaxation | Molt-4 | IC50 (72 h): 0.12 μM | Apoptosis (↓ MMP; ↑ cleaved PARP, ↑ cleaved caspase-3, ↓ p-Akt, ↓ XIAP protein expression) Oxidative stress (↑ ROS; ↓ HIF-1 α, ↓ HO-1, ↑ catalase, ↑ MnSOD, ↓ NOX4, ↑ NOX2 protein expression) ↑ Hsp70 protein expression ↓ EGFR, ↓ p-EGFR, ↓ β-catenin protein expression | [36] |
| Western blotting on Molt-4 cells | 0.1–0.4 μM | ↓ Topo IIα protein expression | K562 | IC50 (72 h): 0.54 μM | Apoptosis (↓ MMP; ↑ cleaved PARP, ↑ cleaved caspase-3, ↓ p-Akt, ↓ XIAP protein expression) Oxidative stress (↑ ROS; ↓ HIF-1 α protein expression) ↓ β-catenin protein expression | |||
| Western blotting on PC-3 cells | 0.8–3.2 μM | ↓ Topo IIα protein expression | PC-3 | IC50 (72 h): 0.58 μM | Apoptosis (↓ MMP; ↑ cleaved PARP, ↑ cleaved caspase-3, ↓ p-Akt, ↓ XIAP ↓ Bcl-2, ↓ p-mTOR protein expression) Oxidative stress (↑ ROS; ↓ HIF-1 α, ↓ HO-1, ↑ catalase, ↑ MnSOD, ↑ NOX4, ↓ NOX2 protein expression) ↓ Hsp90, ↑ Hsp70 protein expression ↓ Colony formation ↓ Cell migration ↓ EMT ↓ EGFR, ↓ p-EGFR, ↓ β-catenin protein expression | |||
| Du145 | IC50 (72 h): 0.33 μM | Apoptosis (↓ MMP; ↑ cleaved PARP, ↑ cleaved caspase-3, ↓ p-Akt, ↓ XIAP ↓ Bcl-2, ↓ p-mTOR protein expression) Oxidative stress (↑ ROS; ↓ HIF-1 α, ↓ HO-1, ↑ catalase, ↑ MnSOD, ↓ NOX4, ↑ NOX2 protein expression) ↓ Hsp90 protein expression ↓ colony formation ↓ cell migration ↓ EMT | ||||||
| CCD966S d | IC50 (72 h): 1.54 μM | |||||||
| NR8383 d | IC50 (72 h): 6.77 μM | |||||||
| Bastadin-14 | Psammaplysilla purpurea | Not indicated | IC50: 2 μg/mL | ↓ Topo IIα protein activity | A-549 | IC50: 2 μg/mL | [79] | |
| HT-29 | ||||||||
| P388 e | ||||||||
| CV-1 d | IC50: 2.5 μg/mL | |||||||
| Batzelline A | Batzella sp. | Cell-free decatenation reaction of kinetoplast DNA | 25 μg/mL (88.45 μM) | ↓ DNA decatenation (58%) | Panc-1 | IC50 (72 h): >17.68 μM | [54] | |
| Ethidium bromide displacement fluorescence assay | 25 μg/mL (88.45 μM) | DNA intercalation (18%) | AsPC-1 | IC50 (72 h): >17.68 μM | ↓ Cell cycle in phase S | |||
| BxPC-3 | IC50 (72 h): >17.68 μM | |||||||
| Mia PaCa2 | ||||||||
| Vero d | ||||||||
| Batzelline B | Batzella sp. | Cell-free decatenation reaction of kinetoplast DNA | 25 μg/mL (93.02 μM) | ↓ DNA decatenation (63%) | Panc-1 | IC50 (72 h): >18.61μM | [54] | |
| Ethidium bromide displacement fluorescence assay | 25 μg/mL (93.02 μM) | DNA intercalation (21%) | AsPC-1 | IC50 (72 h): >18.61μM | ↓ Cell cycle in phase S | |||
| BxPC-3 | IC50 (72 h): >18.61μM | |||||||
| Mia PaCa2 | ||||||||
| Vero d | ||||||||
| Elenic acid | Plakinastrella sp. | Not indicated | IC50: 0.1 μg/mL | ↓ Topo IIα activity | P388 e | IC50 (time not indicated): 5 μg/mL | [80] | |
| A-549 | ||||||||
| MEL-28 | ||||||||
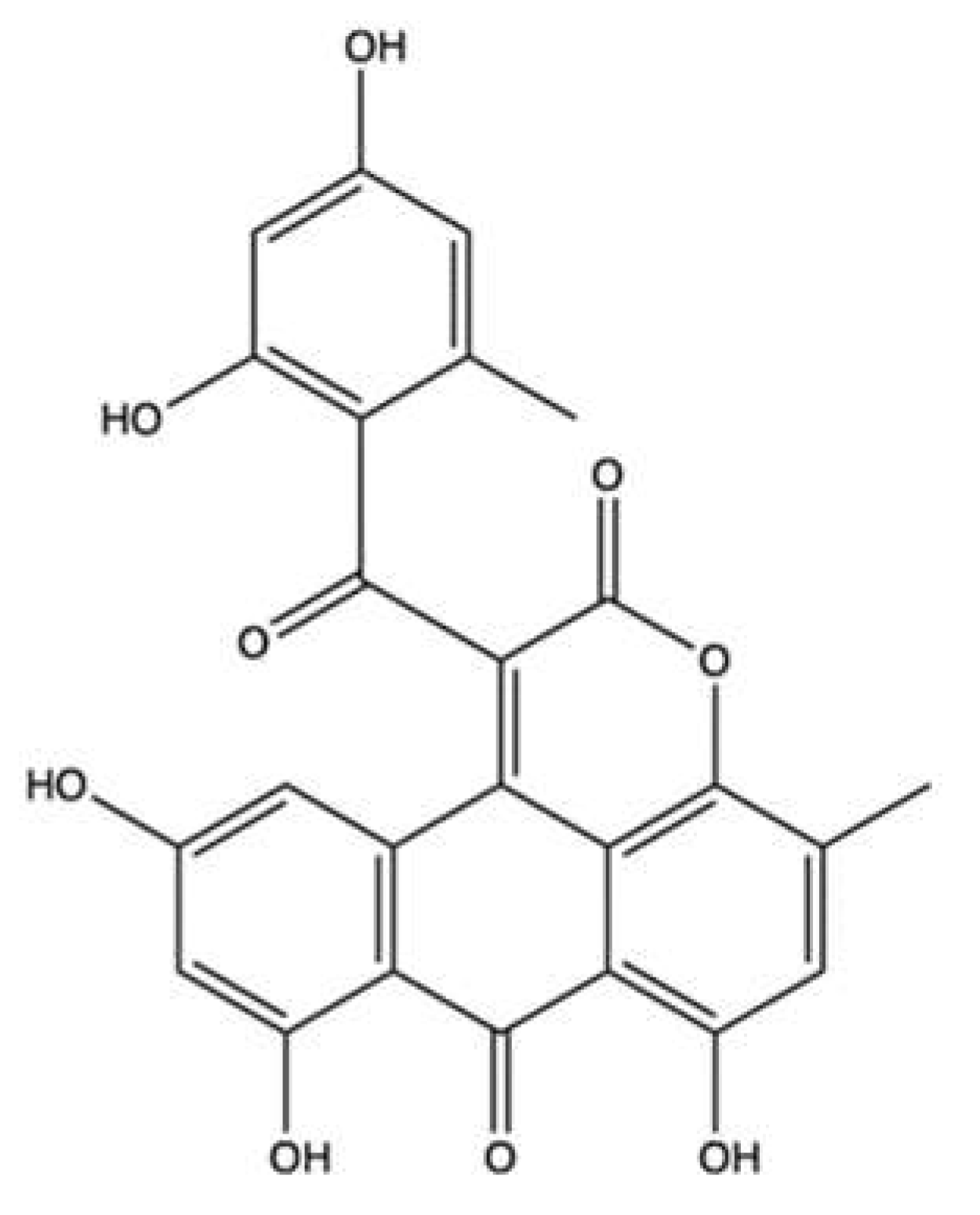
| Halenaquinone | Petrosia sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA and human topo II | IC50: 0.0055 μg/mL (0.017 μM) | ↓ DNA relaxation | Molt-4 | IC50 (24 h): 0.61 μg/mL IC50 (72 h): 0.18 μg/mL | Apoptosis (↓ MMP; ↑ c-PARP, ↑ cleaved caspase-3, ↑ cleaved caspase-7, ↑ cleaved caspase-8, ↑ cleaved caspase-9, ↑ Bax, ↑ cyt c, ↓ Bcl-2, ↓ Bid protein expression) ↓ p-Akt, ↓ p-PTEN, ↓ p-GSK3β, ↓ p-PDK1 and ↓ HKII protein expression Oxidative stress (↑ ROS) Inhibition of HDAC activity (↑ acetyl-H3, ↑ acetyl-H3K18 protein expression) | [75] |
| Western Blotting on Molt-4 cells | 1.25 μg/mL | ↓ Topo IIα protein expression | Immunodeficient athymic mice bearing Molt-4 xenograft | ↓ Tumor growth, ↓ tumor weight, ↓ tumor volume (1μg/g/day for 30 days) | ||||
| K562 | IC50 (72 h): 0.48 μg/mL | Apoptosis (↑ cleaved PARP, ↑ cleaved caspase-3, ↑ cleaved caspase-7 protein expression) | ||||||
| MDA-MB-231 | IC50 (72 h): 8 μg/mL | |||||||
| DLD-1 | IC50 (72 h): 6.76 μg/mL | |||||||
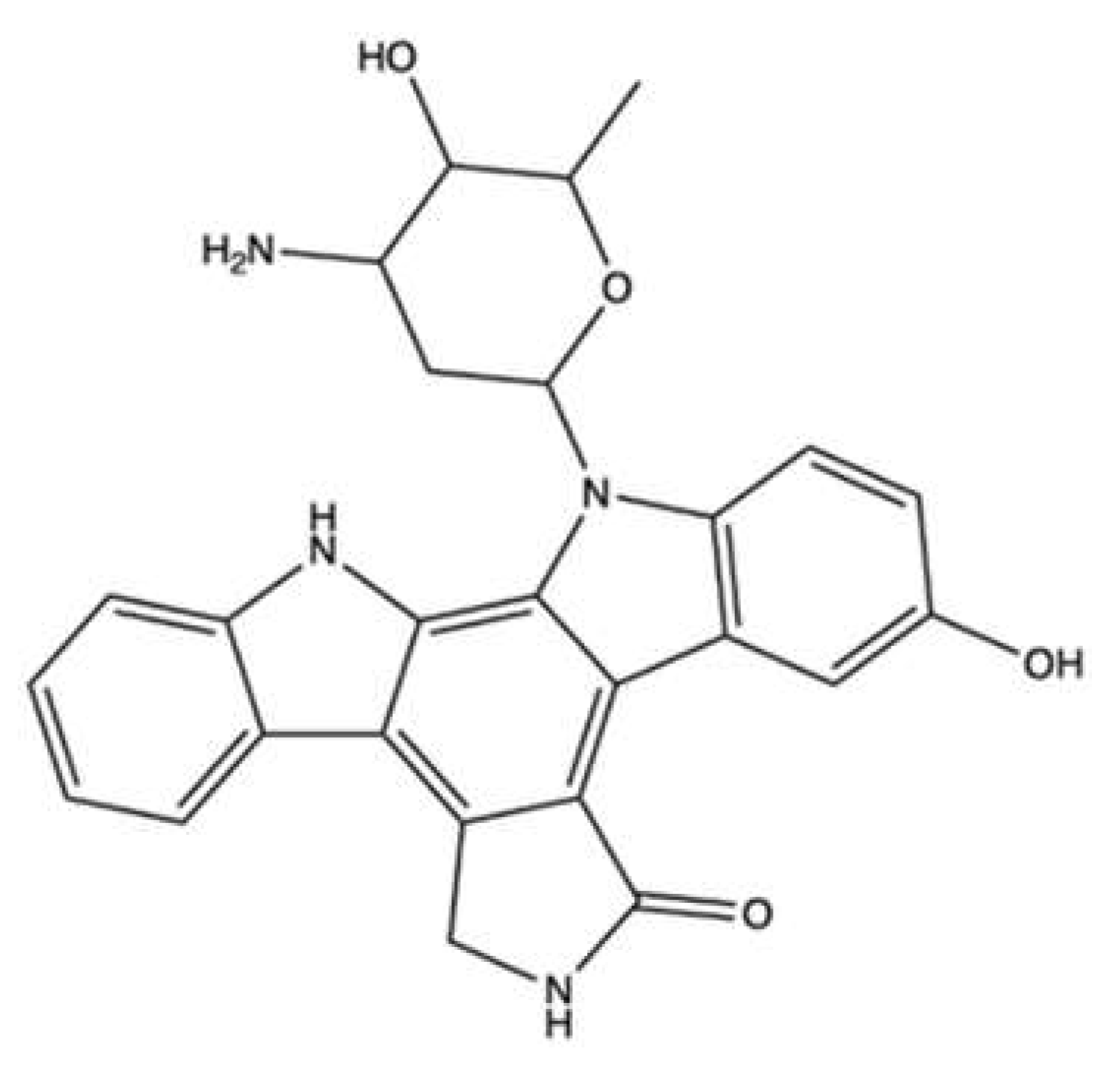
| Heteronemin | Hippospongia sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA and human topo II | 2.56–40.9 μM | ↓ DNA relaxation and formation of supercoiled DNA products in the presence of topo IIα | LNCaP | IC50 (24 h) 1.4 μM IC50 (48 h): 0.8 μM IC50 (72 h): 0.4 μM | Apoptosis (↓ MMP; ↑ cleaved PARP, ↑ cleaved caspase-3 protein expression) Oxidative stress (↑ ROS) ↑ ER stress (↑ Ca2+ release, ↑ IRE 1α, ↑ Bip, ↑ CHOP, ↑ Hsp70, ↓ ATF6, ↓ PERK protein expression) ↓ Hsp90, ↓ IRAK1, ↓ p-Akt, ↓ XIAP, ↓ Rb2, ↓ HDAC1, ↓ PCNA ↓ CDK4, ↓ p-STAT3, ↑ Hsp70 protein expression Autophagy (↑ LC3-II protein expression) | [62] |
| Western blotting on LnCaP cells | 0.64–2.56 μM | ↓ Topo IIα protein expression | PC-3 | IC50 (24 h): 2.7 μM | Apoptosis | |||
| Immunodeficient athymic mice bearing LNcaP xenograft | IC50 (24 h): 7 μM | ↓ Tumor size, ↓ tumor growth (1 mg/Kg b.w.; for 29 days) | ||||||
| Hippospongic acid A | Hippospongia sp. | Cell-free DNA relaxation assay using supercoiled pUC19 DNA plasmid and topo II | IC50: 15 μM | ↓ DNA relaxation | NUGC-3 | IC50 (time not indictaed): 9.5 μM | ↓ Cell cycle in phases G1 and G2/M Apoptosis (↑ DNA fragmentation) | [55] |
| Isobatzelline A | Batzella sp. | Cell-free decatenation reaction of kinetoplast DNA | 25 μg/mL (88.73 μM) | ↓ DNA decatenation (36%) | Panc-1 | IC50 (72 h): 9.37 μM | [54] | |
| Ethidium bromide displacement fluorescence assay | 25 μg/mL (88.73 μM) | DNA intercalation (54%) | AsPC-1 | IC50 (72 h): 1.74 μM | ↓ Cell cycle in phase S | |||
| BxPC-3 | IC50 (72 h): 2.39 μM | |||||||
| Mia PaCa2 | IC50 (72 h): 4.34 μM | |||||||
| Verod | IC50 (72 h): >17.75 μM | |||||||
| Isobatzelline C | Batzella sp. | Cell-free decatenation reaction of kinetoplast DNA | 25 μg/mL (106.38 μM) | ↓ DNA decatenation (27%) | Panc-1 | IC50 (72 h): 9.99 μM | [54] | |
| Ethidium bromide displacement fluorescence assay | 25 μg/mL (106.38 μM) | DNA intercalation (56%) | AsPC-1 | IC50 (72 h): 1.72 μM | ↓ Cell cycle in phase S | |||
| BxPC-3 | IC50 (72 h): 1.31 μM | |||||||
| Mia PaCa2 | IC50 (72 h): 2.34 μM | |||||||
| Verod | IC50 (72 h): >21.28 μM | |||||||
| Isobatzelline D | Batzella sp. | Cell-free decatenation reaction of kinetoplast DNA | 25 μg/mL (89.61 μM) | ↓ DNA decatenation (26%) | Panc-1 | IC50 (72 h): 5.72 μM | [54] | |
| Ethidium bromide displacement fluorescence assay | 25 μg/mL (89.61 μM) | DNA intercalation (47%) | AsPC-1 | IC50 (72 h): 1.48 μM | ↓ cell cycle in phase S | |||
| BxPC-3 | IC50 (72 h): 1.48 μM | |||||||
| Mia PaCa2 | IC50 (72 h): 2.67 μM | |||||||
| Verod | IC50 (72 h): 15.70 μM | |||||||
| Isobatzelline E | Batzella sp. | Cell-free decatenation reaction of kinetoplast DNA | 25 μg/mL (107.30 μM) | ↓ DNA decatenation (95%) | Panc-1 | IC50 (72 h): >21.46 μM | [54] | |
| Ethidium bromide displacement fluorescence assay | 25 μg/mL (107.30 μM) | DNA intercalation (27%) | AsPC-1 | IC50 (72 h): >21.46 μM | ↓ Cell cycle in phase G2 | |||
| BxPC-3 | IC50 (72 h): >21.46 μM | |||||||
| Mia PaCa2 | ||||||||
| Verod | ||||||||
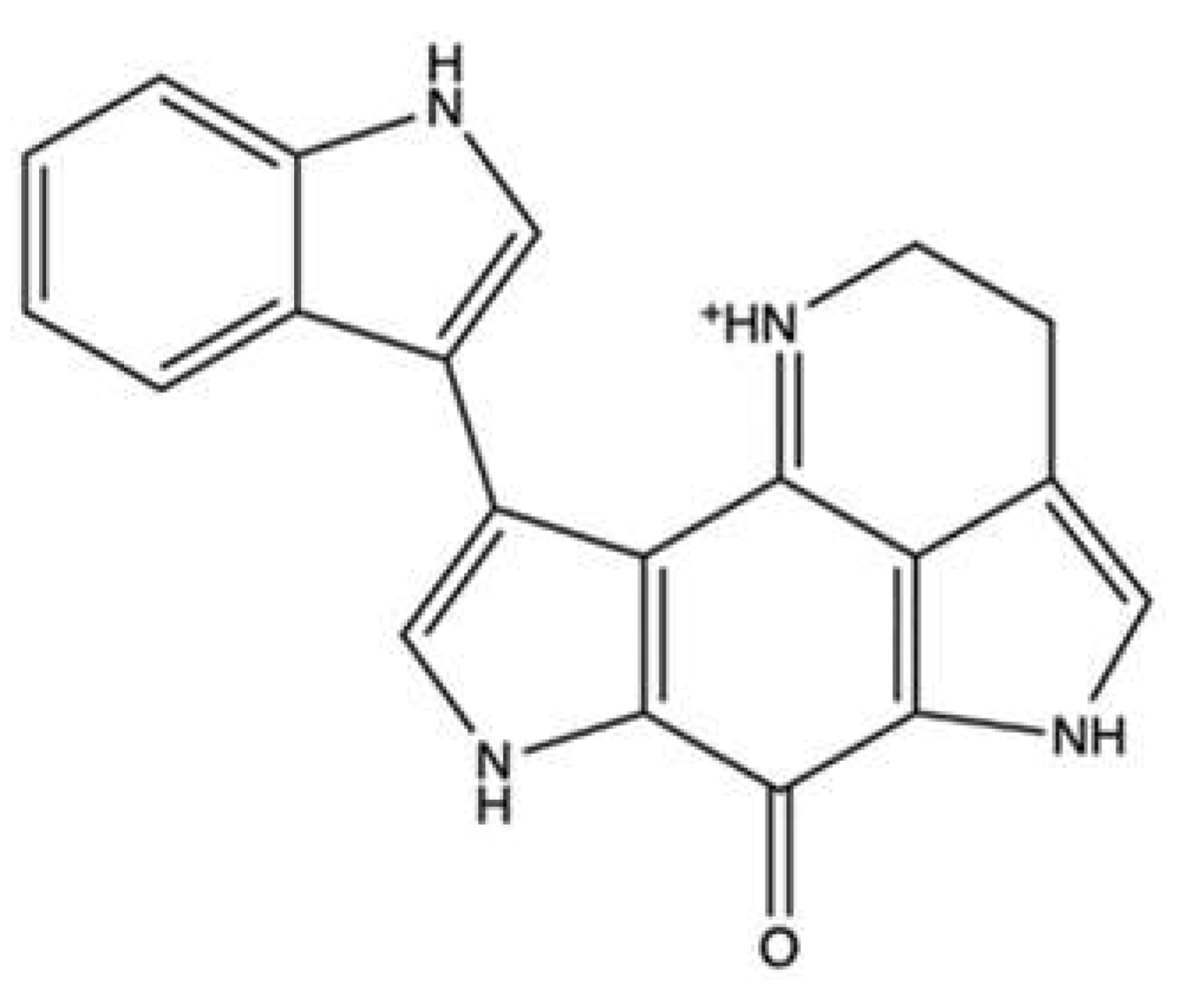
| Makaluvamine A | Zyzzya cf. marsailis | Cell-free decatenation reaction of kinetoplast DNA | IC90: 41 μM | ↓ DNA relaxation | HCT-116 | IC50 (time not indicated): 1.3 μM | [49] | |
| Cell-free DNA cleavage assay with supercoiled pBR322 DNA plasmid | IC50: 2.1 μM | Topo II-mediated cleavage of plasmid DNA | CHO xrs-6 c | IC50 (time not indicated): 0.41 μM | ||||
| Neutral filter elution assay | Not indicated | Production of cleavable complexes (strand scission factor = 1.38) | Balb/C nu/nu athymic mice bearing OVCAR-3 xenograft | ↓ Tumor mass: T/C: 62% (0.5 mg/kg for 4 weeks) | ||||
| Analysis of absorbance spectra of calf thymus DNA | Not indicated | 53% absorption hypochromism | ||||||
| Zyzzya fuliginosa | Cell-free DNA cleavage assay with radiolabeled and supercoiled rf M13 mp19 DNA plasmid | 91 mM | 17% of topo II-mediated cleavage of plasmid DNA (compared to 100% of etoposide) | [51] | ||||
| Makaluvamine B | Zyzzya cf. marsailis | Cell-free decatenation reaction of kinetoplast DNA | IC90: 500 μM | ↓ DNA relaxation | HCT-116 | IC50 (time not indicated): >50 μM | [49] | |
| Cell-free DNA cleavage assay with supercoiled pBR322 DNA plasmid | IC50: 181 μM | Topo II-mediated cleavage of plasmid DNA | CHO xrs-6 c | IC50 (time not indicated): 13.49 μM | ||||
| Makaluvamine C | Zyzzya cf. marsailis | Cell-free decatenation reaction of kinetoplast DNA | IC90: 420 μM | ↓ DNA relaxation | HCT-116 | IC50 (time not indicated): 36.2 μM | / | [49] |
| in vitro cell-free DNA cleavage assay with supercoiled pBR322 DNA plasmid | IC50: 1.2 μM | Topo II-mediated cleavage of plasmid DNA | ||||||
| Analysis of absorbance spectra of calf thymus DNA | Not indicated | 66% absorption hypochromism | CHO xrs-6 c | IC50 (time not indicated): 5.4 μM | ||||
| Balb/C nu/nu athymic mice bearing OVCAR-3 xenograft | ↓ Tumor mass: T/C: 48% (5 mg/kg for 4 weeks) | |||||||
| Immune competent mice inoculated with P388 e cells | ↑ MLS: 18% (5 mg/kg for 4 weeks) | |||||||
| Zyzzya fuliginosa | Cell-free DNA cleavage assay with radiolabeled and supercoiled rf M13 mp19 DNA plasmid and human topo II | 91 mM | 16% of topo II-mediated cleavage of plasmid DNA (compared to 100% of etoposide) | [51] | ||||
| Cell-free cleavage assay of pUC 19 radiolabeled DNA with human topo II | 33–466 μM | Cleavage of pUC 19 DNA at nucleoside A329 Formation of cleavable complex | ||||||
| Makaluvamine D | Zyzzya cf. marsailis | Cell-free decatenation reaction of kinetoplast DNA | IC90: 320 μM | ↓ DNA relaxation | HCT-116 | IC50 (time not indicated): 17.1 μM | [49] | |
| Cell-free DNA cleavage assay with supercoiled pBR322 DNA plasmid | IC50: 52 μM | Topo II-mediated cleavage of plasmid DNA | CHO xrs-6 c | IC50 (time not indicated): 14 μM | ||||
| Zyzzya fuliginosa | Cell-free DNA cleavage assay with radiolabeled and supercoiled rf M13 mp19 DNA plasmid and topo II | 91 mM | 5% of topo II-mediated cleavage of plasmid DNA (compared to 100% of etoposide) | [51] | ||||
| in vitro cell-free cleavage assay of pUC 19 radiolabeled DNA with human topo II | 33–466 μM | Cleavage of pUC 19 DNA at nucleoside A329 Formation of cleavable complex | ||||||
| Makaluvamine E | Zyzzya cf. marsailis | Cell-free decatenation reaction of kinetoplast DNA | IC90: 310 μM | ↓ DNA relaxation | HCT-116 | IC50 (time not indicated): 1.2 μM | [49] | |
| Cell-free DNA cleavage assay with supercoiled pBR322 DNA plasmid | IC50: 15 μM | Topo II-mediated cleavage of plasmid DNA | CHO xrs-6 c | IC50 (time not indicated): 1.7 μM | ||||
| Zyzzya fuliginosa | Cell-free DNA cleavage assay with radiolabeled and supercoiled rf M13 mp19 DNA plasmid and topo II | 91 mM | 22% of topo II-mediated cleavage of plasmid DNA (compared to 100% of etoposide) | [51] | ||||
| Cell-free cleavage assay of pUC 19 radiolabeled DNA with human topo II | 33–466 μM | Cleavage of pUC 19 DNA at nucleoside A329 Formation of cleavable complex | ||||||
| Makaluvamine F | Zyzzya cf. marsailis | Cell-free decatenation reaction of kinetoplast DNA | IC90: 25 μM | ↓ DNA relaxation | HCT-116 | IC50 (time not indicated): 0.17 μM | [49] | |
| Cell-free DNA cleavage assay with supercoiled pBR322 DNA plasmid | IC50: 1.1 μM | Topo II-mediated cleavage of plasmid DNA | CHO xrs-6 c | IC50 (time not indicated): 0.08 μM | ||||
| Makaluvamine H | Zyzzya fuliginosa | Cell-free DNA cleavage assay with radiolabeled and supercoiled rf M13 mp19 DNA plasmid and topo II | 91 mM | 33% of topo II-mediated cleavage of plasmid DNA (compared to 100% of etoposide) | Athymic nude mice bearing KB xenograft | Not indicated | ↓ Tumor growth (22 mg/kg, days 1, 4, and 8 for 28 days) | [51] |
| Makaluvamine I | Zyzzya fuliginosa | Cell-free DNA cleavage assay with radiolabeled and supercoiled rf M13 mp19 DNA plasmid and topo II | 91 mM | 61% of Topo II-mediated cleavage of plasmid DNA (compared to 100% of etoposide) | CHO xrs-6 c | IC50 (time not indicated): 0.4 μM | [51] | |
| CHO AA8 c | IC50 (time not indicated): 2 μM | |||||||
| Athymic nude mice bearing KB xenograft | ↓ Tumor growth (11 mg/kg, days 1, 4, and 8 for 28 days) | |||||||
| Makaluvamine N | Zyzzya fuliginosa | Cell-free DNA relaxation assay using a supercoiled pBR322 DNA plasmid and human topo II | / | ↓ Topo II unwinding (>90% at 5 μg/mL) | HCT-116 | IC50 (72 h): 0.6 μg/mL | [46] | |
| Zyzzya fuliginosa | Cell-free DNA cleavage assay with radiolabeled supercoiled rf M13 mp19 DNA plasmid and topo II | 91 mM | 26% of topo II-mediated cleavage of plasmid DNA (compared to 100% of etoposide) | [51] | ||||
| Makaluvamine V | Zyzzya fuliginosa | Cell-free DNA cleavage assay with radiolabeled supercoiled rf M13 mp19 DNA plasmid and topo II | 91 mM | 2% of topo II-mediated cleavage of plasmid DNA (compared to 100% of etoposide) | [51] | |||
| Manoalide 25-acetals | Hirtios erecta | Not indicated | IC50: 25 μM | ↓ DNA-unknotting activity of calf thymus DNA topo II | CDF1 mice inoculated with P388 e cells | T/C: 150% (1 μg/g) | [60] | |
| Neoamphidemine | Xestospongia sp. | Cell-free decatenation reaction of kinetoplast DNA | 0.5–30 μM | ↓ DNA decatenation | HEK-293 | IC50 (72 h): 0.8 μM | [29] | |
| Malachite green assay with supercoiled DNA, recombinant human topoIIα | 0–10 μM | Competitive inhibition of topo II-mediated ATP hydrolysis | HEK293-Metnase | IC50 (72 h): 0.5 μM | ||||
| Molecular docking | / | Bind to the ATPase site of topo II | ||||||
| Not indicated | Not indicated | Catenation of DNA to high molecular weight complex | CHO AA8 c | IC50 (time not indicated): 2 μg/mL | [27] | |||
| Not indicated | Not indicated | 3% of topo II-mediated DNA cleavage | ||||||
| Cell-free DNA cleavage assay using radiolabeled and supercoiled rf M13 mp19 DNA and human topo II | Not indicated | Catenation of DNA to high molecular weight complex | HCT-116 | IC50 (72 h): 4.5 μM | [25] | |||
| 50 μM | 8.9% of topo II-mediated DNA cleavage | |||||||
| Transmission electron microscopy analysis | Not indicated | Catenation of DNA | SK-mel-5 | IC50 (72 h): 7.6 μM | ||||
| DNA filter-binding assay | 100–600 μM | Catenation of DNA through DNA aggregation | KB | IC50 (72 h): 6 μM | ||||
| MCF-7 | IC50 (72 h): 1.8 μM | |||||||
| A2780 | IC50 (72 h): 0.9 μM | |||||||
| A2780AD | IC50 (72 h): 0.83 μM | |||||||
| CHO AA8 c | IC50 (72 h): 2.5 μM | |||||||
| CHO xrs-6 c | IC50 (72 h): 1.6 μM | |||||||
| Balb/c nu/nu mice bearing HCT-116 xenograft | ↓ Tumor growth (12.5, 25, and 50 mg/kg for 19 days) | |||||||
| Balb/c nu/nu mice bearing KB xenograft | ↓ Tumor growth (50 mg/kg for 19 days) | |||||||
| Popolohuanone E | Dysidea sp. | Not indicated | IC50: 400 μM | ↓ Topo II activity | P388 e | IC50: 20 μg/mL | [81] | |
| HT-29 | IC50: >20 μg/mL | |||||||
| A549 | IC50: 2.5 μg/mL | |||||||
| CV-1 d | IC50: >20 μg/mL | |||||||
| Secobatzelline A | Batzella sp. | Cell-free decatenation reaction of kinetoplast DNA | 25 μg/mL (98.04 μM) | ↓ DNA decatenation (61%) | Panc-1 | IC50 (72 h): 10.39 μM | [54] | |
| Ethidium bromide displacement fluorescence assay | 25 μg/mL (98.04 μM) | DNA intercalation (34%) | AsPC-1 | IC50 (72 h): 3.62 μM | ↓ Cell cycle in phase S | |||
| BxPC-3 | IC50 (72 h): 4.10 μM | |||||||
| Mia PaCa2 | IC50 (72 h): 5.62 μM | |||||||
| Verod | IC50 (72 h): 14.03 μM | |||||||
| Secobatzelline B | Batzella sp. | Cell-free decatenation reaction of kinetoplast DNA | 25 μg/mL (97.66 μM) | ↓ DNA decatenation (13%) | Panc-1 | IC50 (72 h): 17.38 μM | [54] | |
| Ethidium bromide displacement fluorescence assay | 25 μg/mL (97.66 μM) | DNA intercalation (17%) | AsPC-1 | IC50 (72 h): >19.531 μM | ↓ Cell cycle in phase S | |||
| BxPC-3 | IC50 (72 h): >19.53 μM | |||||||
| Mia PaCa2 | ||||||||
| Verod | ||||||||
| Secoadociaquinone A | Xestopongia sp. | Cell-free decatenation reaction of kinetoplast DNA | IC90: 113 μM | ↓ DNA decatenation | HCT-116 | IC50 (18 + 72 h): >143 μM | [77] | |
| CHO xrs-6 c | IC50 (18 + 72 h): >247 μM | |||||||
| Secoadociaquinone B | Xestopongia sp. | Cell-free decatenation reaction of kinetoplast DNA | IC90: 113 μM | ↓ DNA decatenation | HCT-116 | IC50 (18 + 72 h): >143 μM | [77] | |
| CHO xrs-6 c | IC50 (18 + 72 h): >247 μM | |||||||
| Xestoquinone | Petrosia sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively-supercoiled pHOT1 plasmid DNA and topo II | IC50: 0.094 μM | ↓ DNA relaxation | Molt-4 | IC50 (24 h): 2.95 μM | Apoptosis (↓ MMP; ↑ cleaved PARP, ↑ cleaved caspase-3, ↑ cleaved caspase-7, ↑ cleaved caspase-8, ↑ cleaved caspase-9, ↑ Bax, ↑ Bak, ↑ cyt c, ↑ Fas, ↑ TRADD, ↓ Bcl-2, ↓ Bid, ↓ XIAP protein expression ↑ ER stress (↑ Ca2+ release; ↑ CHOP, ↓ IRE 1α, ↓ PERK protein expression) ↓ HDAC activity (↓ HDAC1, ↓ HDAC3, ↓ HDAC4, ↓ HDAC6, ↓ HDAC7, ↓ HDAC8 protein expression) DNA damage (↑ p-Chk1, ↑ p-Chk2, ↑ γH2AX protein expression) Oxidative stress (↑ ROS) Interaction with Hsp90 | [76] |
| Immunodeficient athymic mice bearing Molt-4 xenograft | ↓ Tumor growth, ↓ tumor weight, ↓ tumor volume (1μg/g/day for 50 days) ↓ HDAC1, ↓ HDAC3, ↓ HDAC8 protein expression | |||||||
| Western Blotting on Molt-4 and K562 cells | 7.84 μM | ↓ Topo IIα protein expression | K562 | IC50 (24 h): 6.22 μM | ||||
| Sup-T1 | IC50 (24 h): 8.58 μM | |||||||
| U937 | IC50 (24 h): 11.12 μM | |||||||
| NR8383 d | IC50 (24 h): >30 μM | |||||||
3. Topo II Inhibitors from Marine Fungi and Bacteria
3.1. Leptosin F
3.2. Pericosine A
3.3. Marinactinone B
3.4. Aspergiolide A
3.5. Jadomycin DS
3.6. 2R-Acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1t-cyclohexanol
3.7. Streptomyces sp. VITJS4 Ethyl Acetate Crude Extract
3.8. Sulochrin
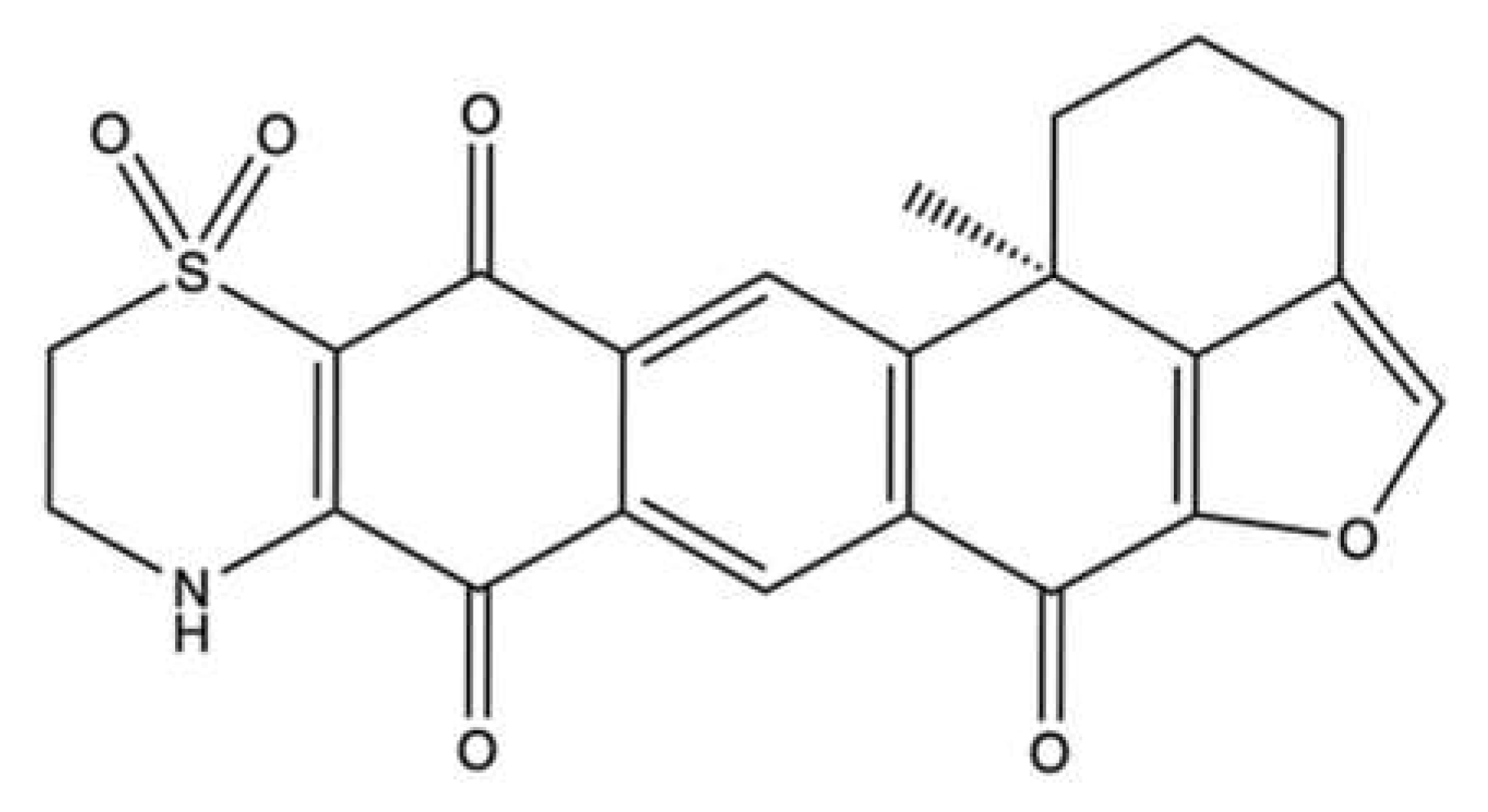
3.9. 3-Hydroxyholyrine A
| Compound | Source | Topo II Inhibitory Activity | Antitumor Effect(s) | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Assay | Concentration(s) Tested or IC50 | Outcomes | Experimental Model | Cytotoxic Activity | Other Antitumor Mechanism(s) | |||
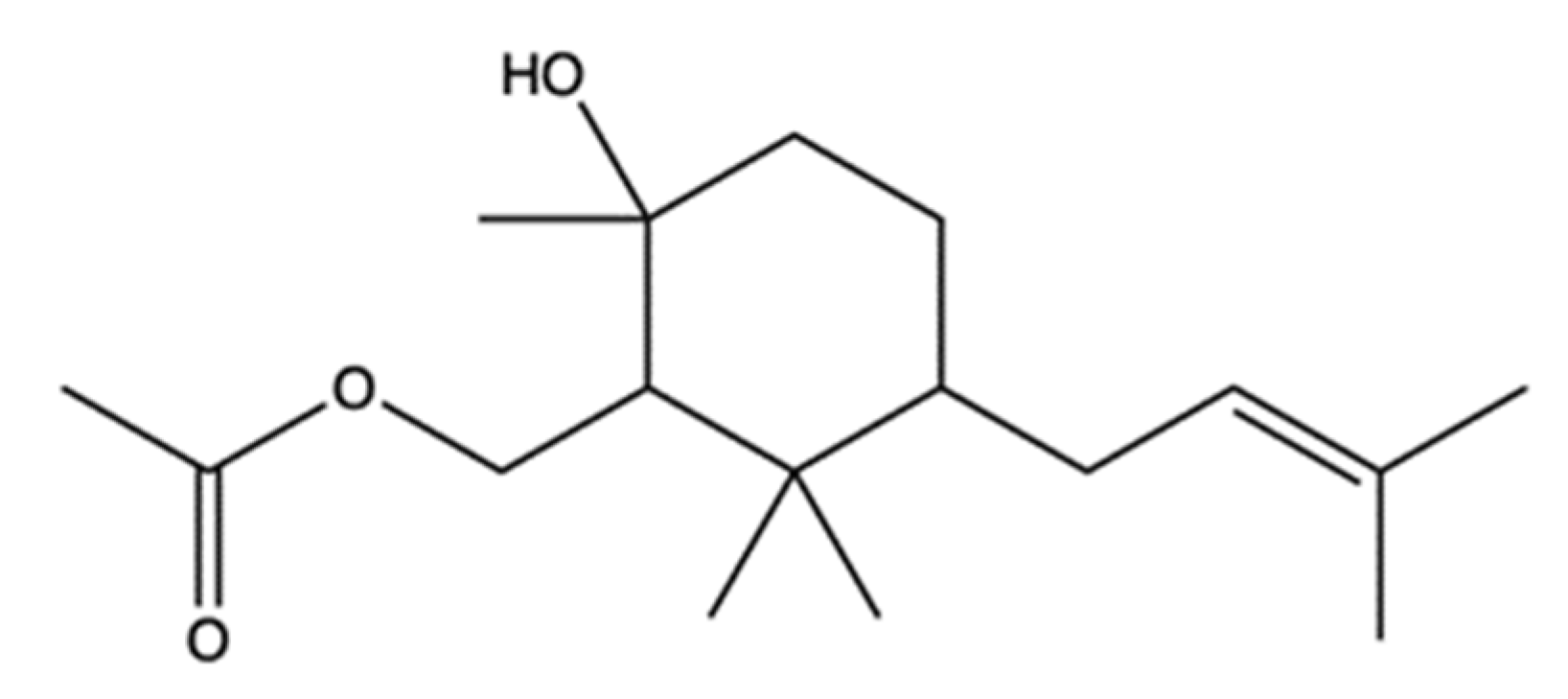
| 2R-acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1t-cyclohexanol | Bacterium Streptomyces sp. VITJS8 | Molecular docking | HepG2 | IC50 (16 h): 250 µg/mL | Apoptosis (caspase-9, caspase-8, caspase-3 cleavage, regulation of Bcl-2 family proteins, cell shrinkage, chromatin condensation, apoptotic bodies, DNA fragmentation, incomplete nuclear membrane) ↓ Cell growth (cell cycle arrest: ↑ cells in S and G2/M phases, ↓ cells in G0/G1 phase) | [94] | ||
| 3-hydroxyholyrine A | Bacterium Streptomyces strain OUCMDZ-3118 | Cell free DNA relaxation assay using supercoiled pBR322 DNA plasmid and topo II α | Not indicated | A-549 | IC50 (48 h): 0.51 µM | [97] | ||
| MCF-7 | IC50 (48 h): 5.0 µM | |||||||
| K562 | IC50 (48 h): 7.2 µM | |||||||
| AGS | IC50 (48 h): 1.7 µM | Apoptosis (↓ survivin protein expression) DNA damage (↑ γ-H2AX protein expression) | ||||||
| MKN45 | IC50 (48 h): 4.3 µM | Apoptosis (↓ survivin protein expression) | ||||||
| Aspergiolide A | Fungus Aspergillus glaucus | A-549 | IC50 (24 h): 0.13 µM | [88] | ||||
| HL-60 | IC50 (72 h): 0.28 µM | |||||||
| BEL-7402 | IC50 (24 h): 7.5 µM | |||||||
| P388 f | IC50 (72 h): 35.0 µM | |||||||
| Spectrofluorimetric decatenation reaction of kinetoplast DNA | 10–100 µM | ↓ Topo II activity | HeLa | IC50 (72 h): 2.37–7.07 µM | [87] | |||
| SMMC-7721 | ||||||||
| SGC-7901 | ||||||||
| MCF-7 | ||||||||
| MDA-MB-468 | ||||||||
| U251 | ||||||||
| A431 | ||||||||
| SK-OV-3 | ||||||||
| BxPC-3 | ||||||||
| 786-O | ||||||||
| BEL-7402 | IC50 (72 h): 2.37–7.07 µM | Apoptosis (procaspase-3, procaspase-8, procaspase-9 and PARP cleavage, ↑ Bax protein expression, ↓ Bcl-2 protein expression) DNA damage (↑ γ-H2AX protein expression) | ||||||
| KN mice inoculated with H22 f cells | ↓ Tumor growth (5, 15, 45 mg/kg i.p.) | |||||||
| Nude mice bearing BEL-7402 xenografts | ↓ Tumor volume (7, 14, 28 mg/kg/day i.p. for 21 days) | |||||||
| Jadomycin DS | Bacterium Streptomyces venezuelae ISP5230 | WaterLOGSY NMR spectroscopy | Interaction with topo IIβ | [91] | ||||
| Leptosin F | Fungus Lestoshaeria sp. | Cell-free decatenation reaction of kinetoplast st DNA | 10–30 µM | ↓ Topo II activity | RPMI8402 | IC50 (72 h): 8.2 nM | Apoptosis (↑ caspase-3 activity, ↑ DNA degradation) | [82] |
| 293 d | Apoptosis (caspase-3 activation Akt inactivation) | |||||||
| P388 f | ED50: 0.056 µg/cm3 | [98] | ||||||
| Marinactinone B | Bacterium Marinactinospora thermotolerans | Cell free relaxation assay using supercoiled pBV220 DNA plasmid and topo II from rat liver cells | 607 µM | ↓ Topo II activity | SW1990 | IC50 (72 h): 99 µM | [86] | |
| Pericosine A | Fungus Periconia byssoides | 100–300 mM | ↓ Topo II activity | P388 f | ED50: 0.1 µg/mL | [83] | ||
| HBC-4 | Log GI50/M: -4.76 | |||||||
| BSY-1 | Log GI50/M: -4.75 | |||||||
| HBC-5 | Log GI50/M: -5.22 | |||||||
| MCF-7 | Log GI50/M: -4.66 | |||||||
| MDA-MB-231 | Log GI50/M: -4.74 | |||||||
| U-251 | Log GI50/M: -4.76 | |||||||
| SF-268 | Log GI50/M: -4.72 | |||||||
| SF-295 | Log GI50/M: -4.62 | |||||||
| SF-539 | Log GI50/M: -4.71 | |||||||
| SNB-75 | Log GI50/M: -7.27 | |||||||
| SNB-78 | Log GI50/M: -4.71 | |||||||
| HCC2998 | Log GI50/M: -4.75 | |||||||
| KM-12 | Log GI50/M: -4.73 | |||||||
| HT-29 | Log GI50/M: -4.70 | |||||||
| WiDr | Log GI50/M: -4.64 | |||||||
| HCT-15 | Log GI50/M: -4.77 | |||||||
| HCT-116 | Log GI50/M: -4.75 | |||||||
| NCI-H23 | Log GI50/M: -4.78 | |||||||
| NCI-H226 | Log GI50/M: -4.80 | |||||||
| NCI-H522 | Log GI50/M: -4.95 | |||||||
| NCI-H460 | Log GI50/M: -4.72 | |||||||
| A-549 | Log GI50/M: -4.61 | |||||||
| DMS273 | Log GI50/M: -4.68 | |||||||
| DMS114 | Log GI50/M: -4.82 | |||||||
| LOX-IMVI | Log GI50/M: -4.72 | |||||||
| OVCAR-3 | Log GI50/M: -4.85 | |||||||
| OVCAR-4 | Log GI50/M: -4.68 | |||||||
| OVCAR-5 | Log GI50/M: -4.79 | |||||||
| OVCAR-8 | Log GI50/M: -4.78 | |||||||
| SK-OV-3 | Log GI50/M: -4.76 | |||||||
| RXF-631L | Log GI50/M: -4.73 | |||||||
| ACHN | Log GI50/M: -4.72 | |||||||
| St-4 | Log GI50/M: -4.65 | |||||||
| MKN1 | Log GI50/M: -4.78 | |||||||
| MKN7 | Log GI50/M: -4.70 | |||||||
| MKN28 | Log GI50/M: -4.72 | |||||||
| MKN45 | Log GI50/M: -4.75 | |||||||
| MKN74 | Log GI50/M: -4.69 | |||||||
| Mice inoculated with P388 f cells | ↑ Survival days compared to controls (25 mg/kg administered i.p. on day 1 and 5) | |||||||
| Streptomyces sp. VITJS4 strain crude extract | Bacterium Streptomyces sp. VITJS4 | Molecular docking | HeLa | IC50 (time not indicated): 50 µg/mL | Apoptosis DNA damage (stained nuclei with round morphology, chromatin condensation, DNA fragmentation) | [95] | ||
| HepG2 | IC50 (time not indicated): 50 µg/mL | |||||||
| Sulochrin | Fungus Aspergillus falconensis | Molecular docking | L5178Y f | IC50 (24 h): 5.1 µM | [96] | |||
| MDA-MB-231 | No cytotoxic activity | ↓ Cell migration | ||||||
4. Topo II Inhibitors Derived from Ascidians, Echinoderms and Marine Microalgae
4.1. Wakayin
4.2. Ascididemin
4.3. Gymnodinium sp. A3 Acidic Polysaccharide
4.4. Echinoside A
4.5. Eusynstyelamide B
| Compounds | Source | Topo II Inhibitory Activity | Antitumor Effect(s) | Ref | ||||
|---|---|---|---|---|---|---|---|---|
| Assay | Concentration(s) Tested or IC50 | Outcomes | Experimental Model | Cytotoxic Activity | Other Antitumor Mechanism(s) | |||
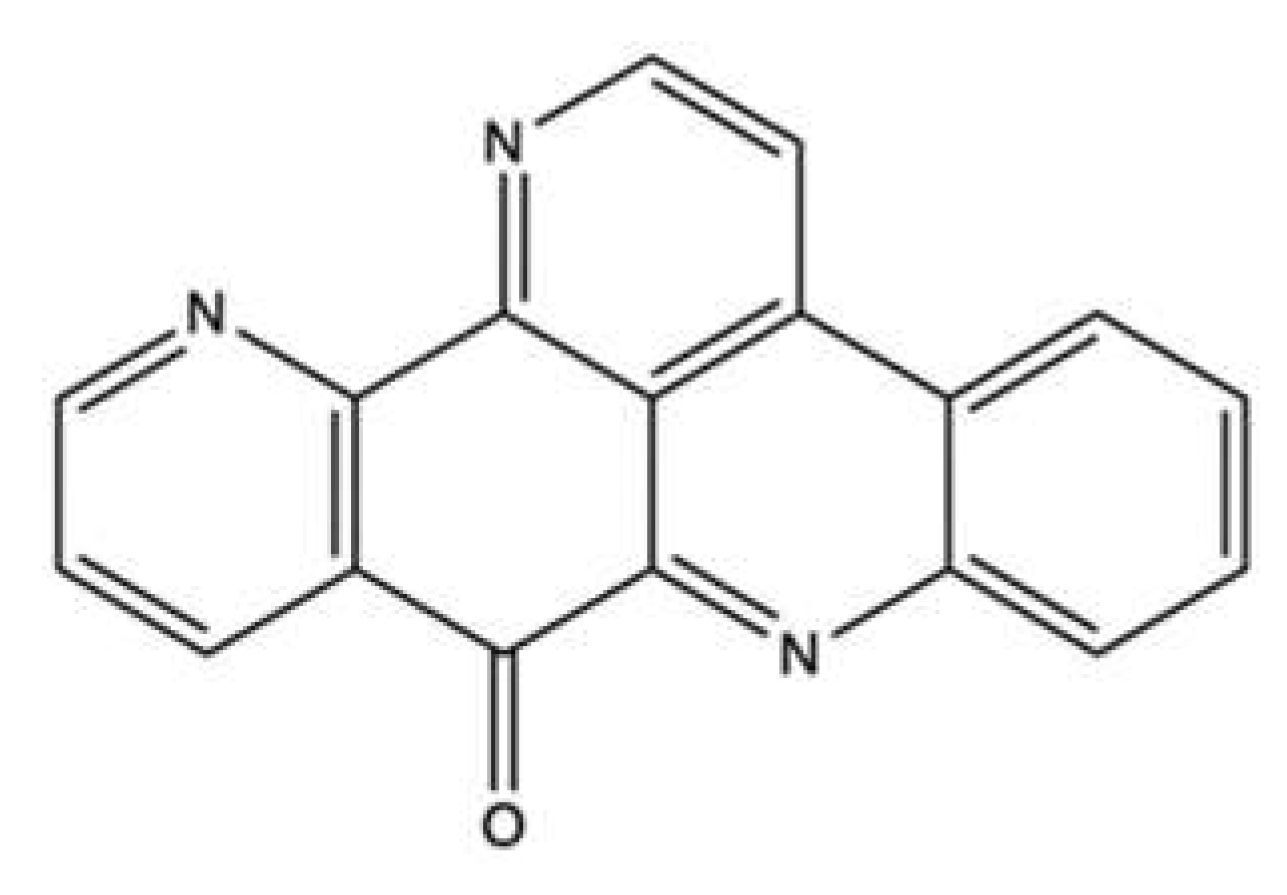
| Ascididemin | Ascidians Cystodytes dellechiajei and Didemnum sp. | L1210 d | IC50 (time not indicated): 0.39 µg/mL | ↑ Ca2+ release in sarcoplasmic reticulum | [102] | |||
| P388 d | IC50 (time not indicated): 0.4 µM | [103] | ||||||
| HCT-116 | IC50 (time not indicated): 0.3 µM | |||||||
| MCF-7 | IC50 (time not indicated): 0.3 µM | |||||||
| CHO xrs-6 b | IC50 (time not indicated): 0.03 µM | |||||||
| CHO EM9 c | IC50 (time not indicated): 0.3 µM | |||||||
| IC50: 30 µM | ↓ Topo II activity | [105] | ||||||
| Cell free DNA relaxation assay using supercoiled pKMp27 DNA plasmid and topo II α | 0.5–50 µM | Weak ↓ topo II activity | HL-60 | IC50 a (72 h): 0.48 µM | Apoptosis (caspase-3 activation, PARP cleavage, DNA fragmentation) Cell cycle inhibition (↑ cells in sub-G1 phase, ↓ cells in G1 and G2 phases) | [104] | ||
| Cell-free DNA cleavage assay using radiolabeled pKMp27 DNA plasmid | 25–100 µM | Stimulation of DNA cleavage Stabilization of DNA–topoisomerase II covalent complexes | HL-60/MX2 e | IC50 a (72 h): 0.65 µM | Cell cycle inhibition (↑ cells in sub-G1 phase, ↓ cells in G1 and G2 phases) | |||
| Cell-free DNA cleavage assay using radiolabeled and supercoiled rf M13 mp19 DNA | 91 µM | ↓ Topo II activity | CHO AA8 | IC50 (72 h): 3.1 µM | [106] | |||
| CHO EM9 c | IC50 (72 h): 0.4 µM | |||||||
| CHO xrs-6 b | IC50 (72 h): 0.7 µM | |||||||
| HCT-116 | IC50 (72 h): 0.1 µM | |||||||
| KB | IC50 (72 h): 0.6 µM | |||||||
| P388 d | IC50 (time not indicated): 0.4 µM | [115] | ||||||
| Bengacarboline | Ascidian Didemnum sp. | 32 µM | ↓ Topo II activity | [116] | ||||
| A-549 | IC50 (72 h): 7.1 µM | [117] | ||||||
| BxPC3 | IC50 (72 h): 10.0 µM | |||||||
| LoVo | IC50 (72 h): 9.9 µM | |||||||
| MCF-7 | IC50 (72 h): 8.6 µM | |||||||
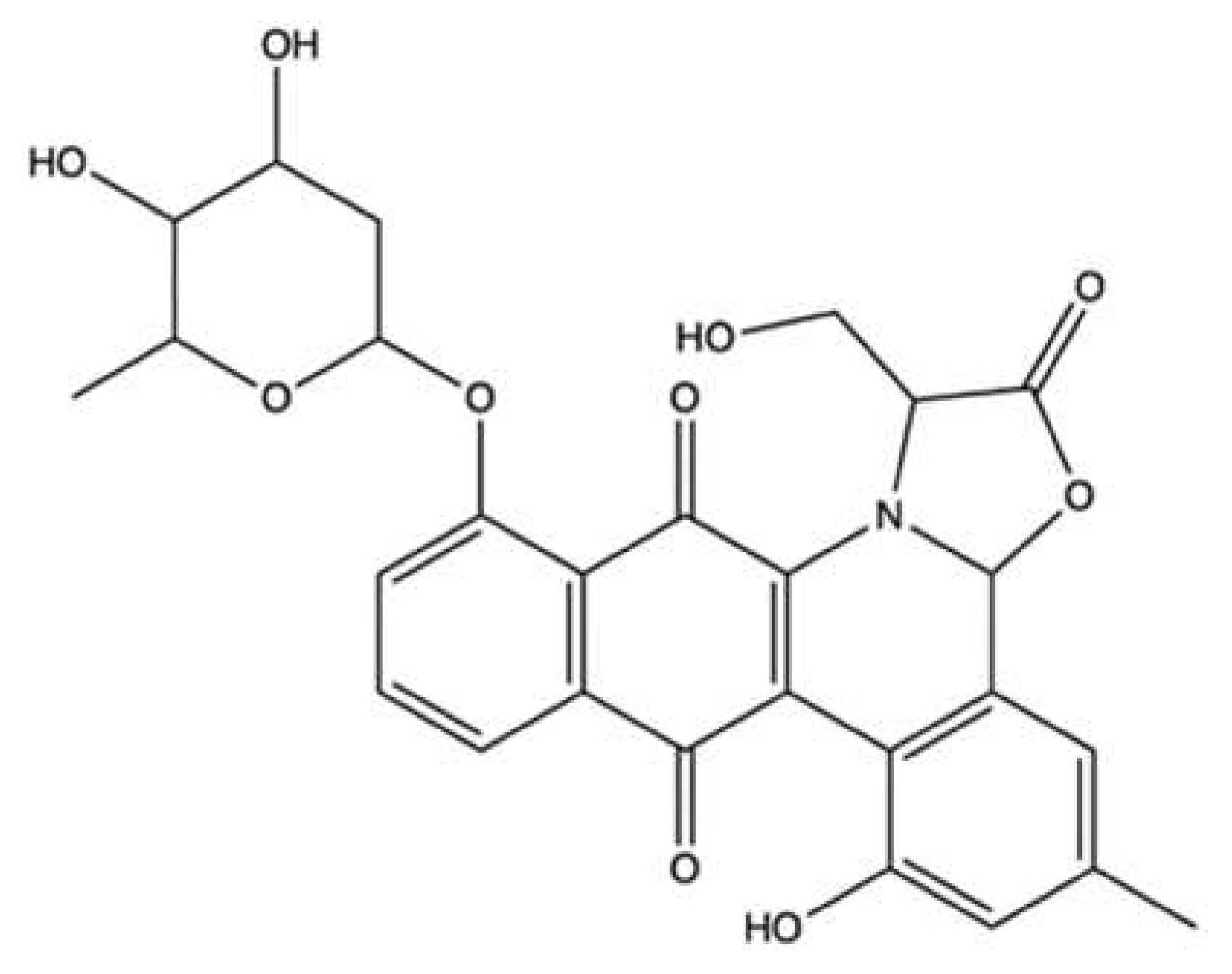
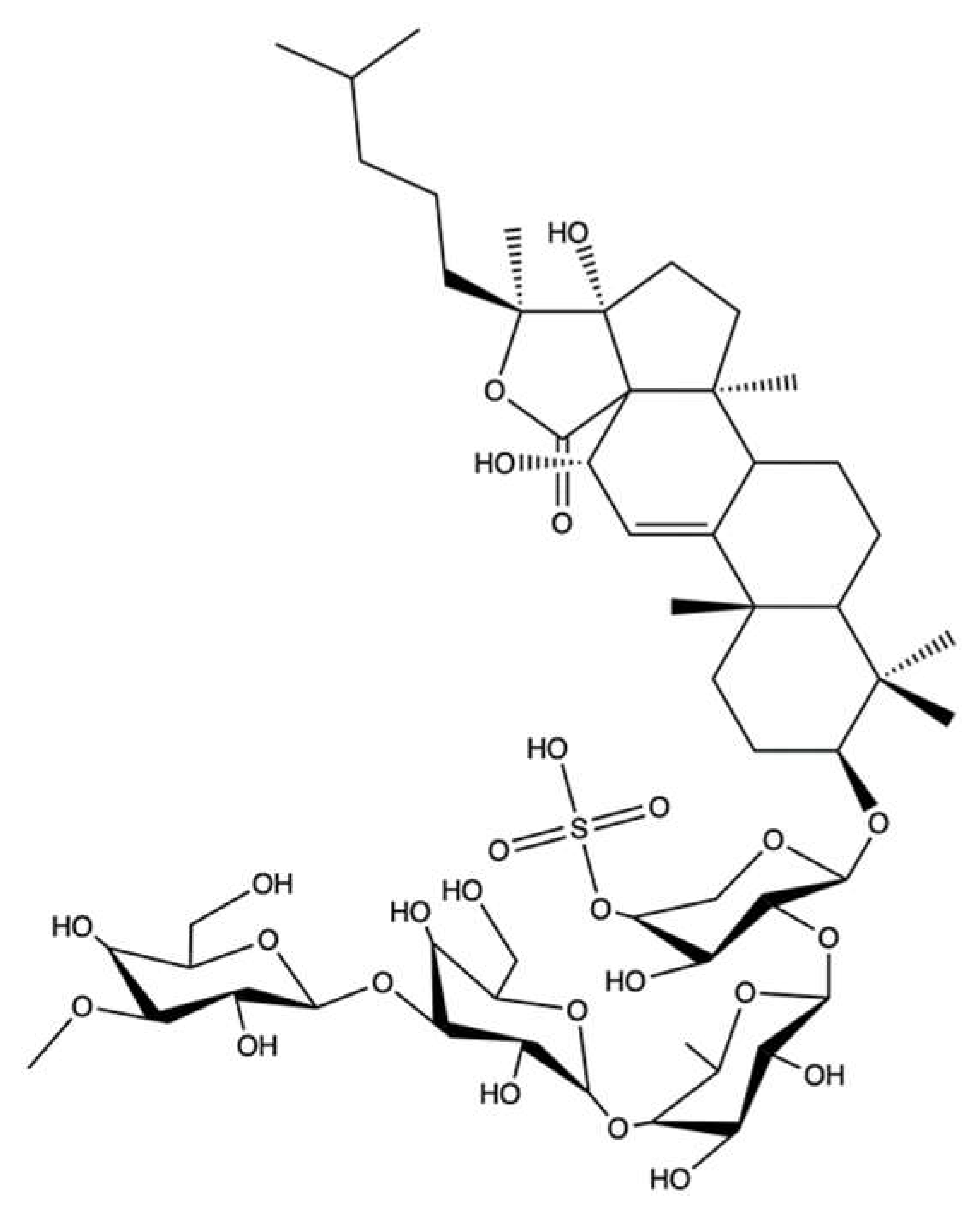
| Echinoside A | Echinoderm Holothuria nobilis | Cell-free decatenation reaction of kinetoplast DNA with topo IIα | 1–125 µM | ↓ DNA decatenation | K562 | IC50 (time not indicated): 5.42 µM | [109] | |
| K562/A02 e | IC50 (time not indicated): 5.31 µM | |||||||
| MCF-7 | IC50 (time not indicated): 1.32 µM | |||||||
| MCF-7/ADRe | IC50 (time not indicated): 1.26 µM | |||||||
| KB | IC50 (time not indicated): 2.78 µM | |||||||
| KB/VCRe | IC50 (time not indicated): 3.29 µM | |||||||
| In vivo complexes of the enzyme (ICE) bioassay | 0.5–1 μM | Formation of stable cleavage complexes | HL-60 | Not indicated | Apoptosis (pro-caspase-3 and pro-caspase-8 cleavage, PARP degradation) DNA damage (↑ γ-H2AX protein expression) | |||
| Cell-free DNA relaxation assay using supercoiled pBR322 plasmid DNA and topo IIα | 5–125 µM | ↓ DNA relaxation | KM mice inoculated with S-180 d cells | ↓ Tumor growth (1.5, 3.0 mg/kg/day for 7 days i.v.) | ||||
| Cell-free DNA unwinding assay using supercoiled pBR322 plasmid DNA and topo IIα | 5–125 µM | No intercalation in the DNA | KM mice inoculated with H22 d cells | ↓ Tumor growth (1.5, 3.0, 6.0 mg/kg/day for 7 days i.v) | ||||
| Ethidium bromide displacement fluorescence assay with human topo IIα | 1–125 µM | No intercalation in the DNA | Nude mice bearing human prostate carcinoma PC-3 xenografts | ↓ Tumor growth (2.6, 5.2 mg/kg/week for 4 weeks i.v.) | ||||
| Cell-free topo IIα –mediated DNA religation assay using supercoiled pBR322 plasmid DNA | ↑ Topo IIα-dependent DSBs formation | |||||||
| Cell free ATP hydrolysis assay with human topo IIα assay | 100 µM | No alteration of the ATPase activity of topo IIα | Nude mice bearing human prostate carcinoma PC-3 xenografts | ↓ Tumor growth (2.6, 5.2 mg/kg/week for 4 weeks i.v.) | ||||
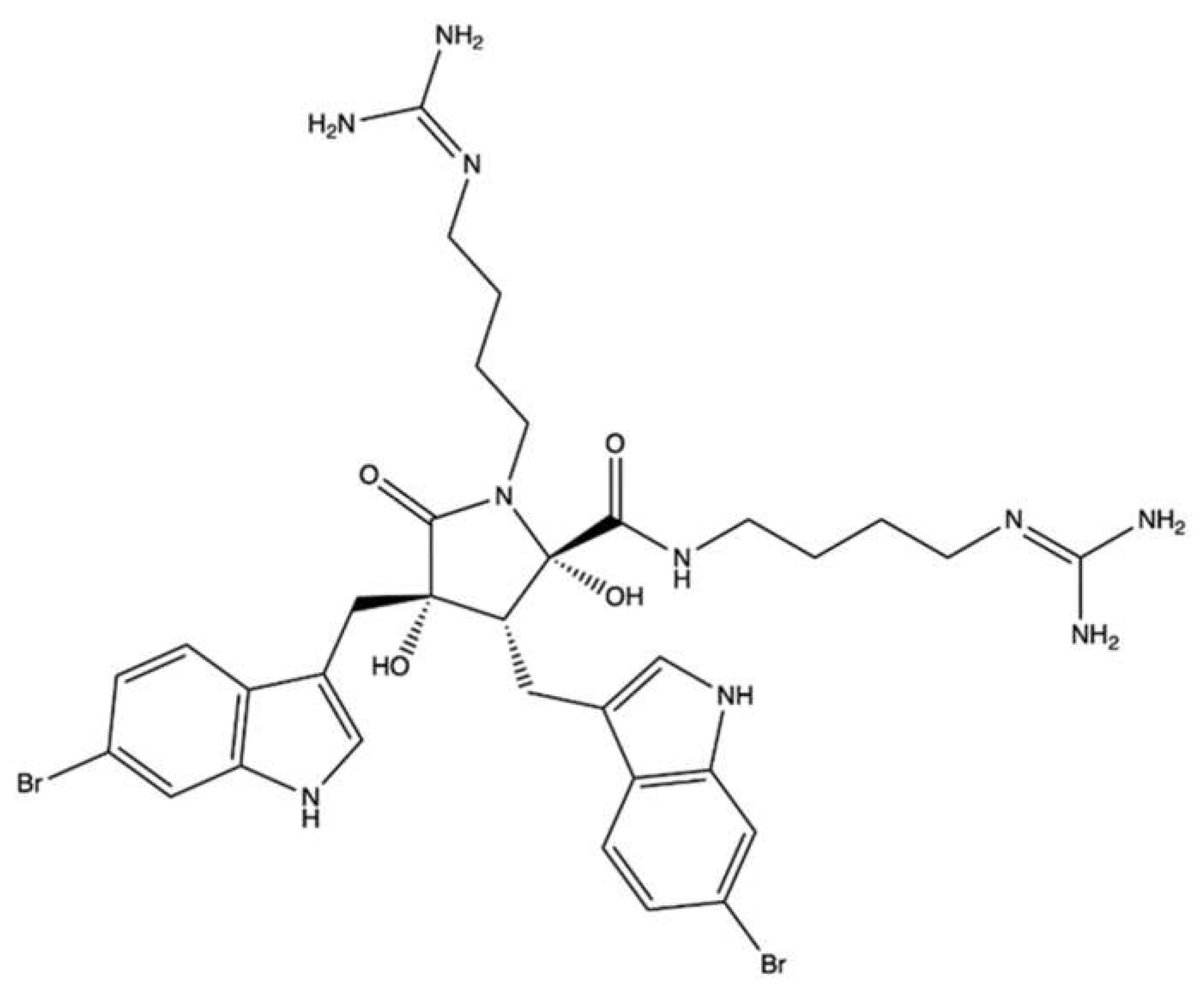
| Eusynstyelamide B | Ascidian Didemnum candidum | MDA-MB-231 | IC50 (72 h): 4.95 µM | Apoptosis (PARP cleavage) Cell cycle inhibition (↑ cells in G2/M phase, ↓ cells in G0/G1 phase) | [112] | |||
| LNCaP | IC50 (72 h): 5.0 µM | Cell cycle inhibition (↑ cells in G2/M phase, ↓ S phase progression, ↓ CDK1, CCNB1, ↓ CDC25A, ↓ CDC2 gene expression, ↑ CDKN1A gene expression, ↑ p21CIP1/WAF1, ↓ total CDC2 protein expression DNA damage induction (↑ γH2AX foci, ↓ expression of MKI67, ↑ GADD45A, GADD45G ↑ CHK2 phosphorylation | [113] | |||||
| Ethidium bromide displacement fluorescence assay with topo II | 6.25–50 µM | No intercalation | MDA-MB-231 | Cell cycle inhibition (↑ cells in G2/M phase, ↓ cells in G0/G1 phase, ↑ CDKN1A gene expression ↑ p21CIP1/WAF1 ↓ CCNB1 gene expression, ↑ total CDC2 protein, ↑ p53, ↑ total p53 expression) DNA damage (↑ γH2AX foci, ↑ CHK2 phosphorylation ↑ CHK1 phosphorylation) | ||||
| DNA melting temperature analysis | 6.25–50 µM | No direct interaction with DNA | ||||||
| Decatenation reaction of kinetoplast DNA with topo II | 25–100 µM | ↓ Decatenation of kDNA | ||||||
| GA3Pl+ | Microalga Gymnodinium sp. A3 | Cell-free decatenation reaction of kinetoplast DNA with recombinant human topo IIα | IC50: 0.017 μg/mL | ↓ Topo IIα activity | HBC-4 | GI50 (time not indicated): 5.2 µg/mL | [108] | |
| Cell-free DNA cleavage assay using supercoiled pT2GN plasmid DNA and recombinant human topo IIα | IC50 0.048 μg/mL | ↓ Topo IIα activity | BSY-1 | GI50 (time not indicated): 0.67 µg/mL | ||||
| HBC-5 | GI50 (time not indicated): 6.2 µg/mL | |||||||
| MCF-7 | GI50 (time not indicated): 2.9 µg/mL | |||||||
| MDA-MB-231 | GI50 (time not indicated): 1.5 µg/mL | |||||||
| U251 | GI50 (time not indicated): 2.0 µg/mL | |||||||
| SF-268 | GI50 (time not indicated): 4.7 µg/mL | |||||||
| SF-295 | GI50 (time not indicated): 2.7 µg/mL | |||||||
| SF-539 | GI50 (time not indicated): 1.8 µg/mL | |||||||
| SNB-75 | GI50 (time not indicated): 1.5 µg/mL | |||||||
| SNB-78 | GI50 (time not indicated): 2.4 µg/mL | |||||||
| HCC2998 | GI50 (time not indicated): 2.3 µg/mL | |||||||
| KM-12 | GI50 (time not indicated): 3.7 µg/mL | |||||||
| HT-29 | GI50 (time not indicated): 3.6 µg/mL | |||||||
| WiDr | GI50 (time not indicated): 3.1 µg/mL | |||||||
| HCT-15 | GI50 (time not indicated): 3.4 µg/mL | |||||||
| HCT-116 | GI50 (time not indicated): 3.7 µg/mL | |||||||
| NCI-H23 | GI50 (time not indicated): 2.8 µg/mL | |||||||
| NCI-H226 | GI50 (time not indicated): 2.2 µg/mL | |||||||
| NCI-H522 | GI50 (time not indicated): 1.3 µg/mL | |||||||
| NCI-H460 | GI50 (time not indicated): 3.8 µg/mL | |||||||
| A-549 | GI50 (time not indicated): 11 µg/mL | |||||||
| DMS273 | GI50 (time not indicated): 2.0 µg/mL | |||||||
| DMS114 | GI50 (time not indicated): 2.7 µg/mL | |||||||
| LOX-IMVI | GI50 (time not indicated): 6.3 µg/mL | |||||||
| OVCAR-3 | GI50 (time not indicated): 2.2 µg/mL | |||||||
| OVCAR-4 | GI50 (time not indicated): 3.2 µg/mL | |||||||
| OVCAR-5 | GI50 (time not indicated): 6.8 µg/mL | |||||||
| OVCAR-8 | GI50 (time not indicated): 4.1 µg/mL | |||||||
| SK-OV-3 | GI50 (time not indicated): 8.1 µg/mL | |||||||
| RXF-631L | GI50 (time not indicated): 9.1 µg/mL | |||||||
| ACHN | GI50 (time not indicated): 8.3 µg/mL | |||||||
| St-4 | GI50 (time not indicated): 8.4 µg/mL | |||||||
| MKN1 | GI50 (time not indicated): 3.0 µg/mL | |||||||
| MKN7 | GI50 (time not indicated): 5.9 µg/mL | |||||||
| MKN28 | GI50 (time not indicated): 7.0 µg/mL | |||||||
| MKN45 | GI50 (time not indicated) 2.9 µg/mL | |||||||
| MKN74 | GI50 (time not indicated): 4.6 µg/mL | |||||||
| GA3Pl- | Microalga Gymnodinium sp. A3 | Decatenation reaction of kinetoplast DNA with recombinant human topo IIα | IC50 0.015 μg/mL | ↓ Topo IIα activity | ||||
| Cell-free DNA cleavage assay using supercoiled pT2GN plasmid DNA and recombinant human topo IIα | IC50 0.052 μg/mL | |||||||
| Meridine | Ascidian Amphicarpa meridiana | Decatenation reaction of kinetoplast DNA | IC50 3 µM | ↓ Topo II activity | P388 d | IC50 (72 h): 0.08 µM | [118] | |
| A-549 | IC50 (72 h): 0.08 µM | |||||||
| HT-29 | IC50 (72 h): 0.84 µM | |||||||
| MEL-28 | IC50 (72 h): 0.08 µM | |||||||
| 75 µM | ↓ Topo II activity | [105] | ||||||
| Wakayin | Ascidian Clavelina sp. | HCT-116 | IC50 (time not indicated): 0.5 µg/mL | [99] | ||||
| Decatenation reaction of kinetoplast DNA with avian topo II | 40–133 µg/mL | ↓ Topo II activity | CHO BR1 | IC50 (72 h): 3.05 µg/mL | [100] | |||
| CHO xrs-6 b | IC50 (72 h): 0.31 µg/mL | |||||||
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Global Cancer Observatory. Available online: https://gco.iarc.fr/ (accessed on 13 September 2022).
- Dias, D.A.; Urban, S.; Roessner, U. A Historical Overview of Natural Products in Drug Discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fimognari, C.; Lenzi, M.; Ferruzzi, L.; Turrini, E.; Scartezzini, P.; Poli, F.; Gotti, R.; Guerrini, A.; Carulli, G.; Ottaviano, V.; et al. Mitochondrial Pathway Mediates the Antileukemic Effects of Hemidesmus Indicus, a Promising Botanical Drug. PLoS ONE 2011, 6, e21544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenzi, M.; Malaguti, M.; Cocchi, V.; Hrelia, S.; Hrelia, P. Castanea sativa Mill. Bark Extract Exhibits Chemopreventive Properties Triggering Extrinsic Apoptotic Pathway in Jurkat Cells. BMC Complement. Altern. Med. 2017, 17, 251. [Google Scholar] [CrossRef]
- Lenzi, M.; Cocchi, V.; Malaguti, M.; Barbalace, M.C.; Marchionni, S.; Hrelia, S.; Hrelia, P. 6-(Methylsulfonyl) Hexyl Isothiocyanate as Potential Chemopreventive Agent: Molecular and Cellular Profile in Leukaemia Cell Lines. Oncotarget 2017, 8, 111697–111714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Scotti, L.; Bezerra Mendonca, F.J.; Ribeiro, F.F.; Tavares, J.F.; da Silva, M.S.; Barbosa Filho, J.M.; Scotti, M.T. Natural Product Inhibitors of Topoisomerases: Review and Docking Study. Curr. Protein Pept. Sci. 2018, 19, 275–291. [Google Scholar] [CrossRef]
- Schirrmacher, V. From Chemotherapy to Biological Therapy: A Review of Novel Concepts to Reduce the Side Effects of Systemic Cancer Treatment. Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef]
- Vann, K.R.; Oviatt, A.A.; Osheroff, N. Topoisomerase II Poisons: Converting Essential Enzymes into Molecular Scissors. Biochemistry 2021, 60, 1630–1641. [Google Scholar] [CrossRef]
- Vélez-Cruz, R.; Osheroff, N. DNA Topoisomerases: Type II. In Encyclopedia of Biological Chemistry; Elsevier: Amsterdam, The Netherlands, 2004; pp. 806–811. ISBN 978-0-12-443710-4. [Google Scholar]
- Buzun, K.; Bielawska, A.; Bielawski, K.; Gornowicz, A. DNA Topoisomerases as Molecular Targets for Anticancer Drugs. J. Enzyme Inhib. Med. Chem. 2020, 35, 1781–1799. [Google Scholar] [CrossRef]
- Bax, B.D.; Murshudov, G.; Maxwell, A.; Germe, T. DNA Topoisomerase Inhibitors: Trapping a DNA-Cleaving Machine in Motion. J. Mol. Biol. 2019, 431, 3427–3449. [Google Scholar] [CrossRef]
- Ormrod, D.; Holm, K.; Goa, K.; Spencer, C. Epirubicin: A Review of Its Efficacy as Adjuvant Therapy and in the Treatment of Metastatic Disease in Breast Cancer. Drugs Aging 1999, 15, 389–416. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, D.; Parihar, L.; Sharma, P.; Sharma, S. A Review on Doxorubicn Induced Cardiotoxicity and Its Molecular Mechanism. Int. J. Pharm. Sci. 2020, 11, 2521–2527. [Google Scholar]
- Singal, P.K.; Iliskovic, N. Doxorubicin-Induced Cardiomyopathy. N. Engl. J. Med. 1998, 339, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Holthuis, J.J.M. Etoposide and Teniposide: Bioanalysis, Metabolism and Clinical Pharmacokinetics. Pharm. Weekbl. 1988, 10, 101–116. [Google Scholar] [CrossRef]
- Lee, J.H.; Wendorff, T.J.; Berger, J.M. Resveratrol: A Novel Type of Topoisomerase II Inhibitor. J. Biol. Chem. 2017, 292, 21011–21022. [Google Scholar] [CrossRef] [Green Version]
- Kaur, R.; Manjal, S.K.; Rawal, R.K.; Kumar, K. Recent Synthetic and Medicinal Perspectives of Tryptanthrin. Bioorg. Med. Chem. 2017, 25, 4533–4552. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Demchuk, O.M. New Perspectives for Fisetin. Front. Chem. 2019, 7, 697. [Google Scholar] [CrossRef]
- Semwal, D.; Semwal, R.; Combrinck, S.; Viljoen, A. Myricetin: A Dietary Molecule with Diverse Biological Activities. Nutrients 2016, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.; Oh, S.H.; Yun, J.H.; Jho, E.H.; Kang, J.-H.; Batsuren, D.; Tunsag, J.; Park, K.H.; Kim, M.; Nho, C.W. A Novel Topoisomerase Inhibitor, Daurinol, Suppresses Growth of HCT116 Cells with Low Hematological Toxicity Compared to Etoposide. Neoplasia 2011, 13, 1043–1057, IN26–IN30. [Google Scholar] [CrossRef] [Green Version]
- Saeed, A.F.U.H.; Su, J.; Ouyang, S. Marine-Derived Drugs: Recent Advances in Cancer Therapy and Immune Signaling. Biomed. Pharmacother. 2021, 134, 111091. [Google Scholar] [CrossRef]
- Demain, A.L.; Fang, A. The Natural Functions of Secondary Metabolites. In History of Modern Biotechnology I; Fiechter, A., Ed.; Advances in Biochemical Engineering/Biotechnology; Springer: Berlin/Heidelberg, Germany, 2000; Volume 69, pp. 1–39. ISBN 978-3-540-67793-2. [Google Scholar]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug Development from Marine Natural Products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Marshall, K.M.; Matsumoto, S.S.; Holden, J.A.; Concepción, G.P.; Tasdemir, D.; Ireland, C.M.; Barrows, L.R. The Anti-Neoplastic and Novel Topoisomerase II-Mediated Cytotoxicity of Neoamphimedine, a Marine Pyridoacridine. Biochem. Pharmacol. 2003, 66, 447–458. [Google Scholar] [CrossRef]
- Li, L.; Abraham, A.D.; Zhou, Q.; Ali, H.; O’Brien, J.V.; Hamill, B.D.; Arcaroli, J.J.; Messersmith, W.A.; LaBarbera, D.V. An Improved High Yield Total Synthesis and Cytotoxicity Study of the Marine Alkaloid Neoamphimedine: An ATP-Competitive Inhibitor of Topoisomerase IIα and Potent Anticancer Agent. Mar. Drugs 2014, 12, 4833–4850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Guzman, F.S.; Carte, B.; Troupe, N.; Faulkner, D.J.; Harper, M.K.; Concepcion, G.P.; Mangalindan, G.C.; Matsumoto, S.S.; Barrows, L.R.; Ireland, C.M. Neoamphimedine: A New Pyridoacridine Topoisomerase II Inhibitor Which Catenates DNA. J. Org. Chem. 1999, 64, 1400–1402. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Caldecott, K.; Pidsley, S.; Banks, G.R. Sensitivity of Chinese Hamster Ovary Mutants Defective in DNA Double Strand Break Repair to Topoisomerase II Inhibitors. Cancer Res. 1989, 49, 7057–7063. [Google Scholar]
- Ponder, J.; Yoo, B.H.; Abraham, A.D.; Li, Q.; Ashley, A.K.; Amerin, C.L.; Zhou, Q.; Reid, B.G.; Reigan, P.; Hromas, R.; et al. Neoamphimedine Circumvents Metnase-Enhanced DNA Topoisomerase IIα Activity through ATP-Competitive Inhibition. Mar. Drugs 2011, 9, 2397–2408. [Google Scholar] [CrossRef] [Green Version]
- Wray, J.; Williamson, E.A.; Royce, M.; Shaheen, M.; Beck, B.D.; Lee, S.-H.; Nickoloff, J.A.; Hromas, R. Metnase Mediates Resistance to Topoisomerase II Inhibitors in Breast Cancer Cells. PLoS ONE 2009, 4, e5323. [Google Scholar] [CrossRef] [Green Version]
- Wray, J.; Williamson, E.A.; Sheema, S.; Lee, S.-H.; Libby, E.; Willman, C.L.; Nickoloff, J.A.; Hromas, R. Metnase Mediates Chromosome Decatenation in Acute Leukemia Cells. Blood 2009, 114, 1852–1858. [Google Scholar] [CrossRef]
- Zhou, Q.; Abraham, A.D.; Li, L.; Babalmorad, A.; Bagby, S.; Arcaroli, J.J.; Hansen, R.J.; Valeriote, F.A.; Gustafson, D.L.; Schaack, J.; et al. Topoisomerase IIα Mediates TCF-Dependent Epithelial-Mesenchymal Transition in Colon Cancer. Oncogene 2016, 35, 4990–4999. [Google Scholar] [CrossRef] [Green Version]
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 499. [Google Scholar] [CrossRef]
- Bian, J.; Dannappel, M.; Wan, C.; Firestein, R. Transcriptional Regulation of Wnt/β-Catenin Pathway in Colorectal Cancer. Cells 2020, 9, 2125. [Google Scholar] [CrossRef] [PubMed]
- Su, J.-H.; Chen, Y.-C.; El-Shazly, M.; Du, Y.-C.; Su, C.-W.; Tsao, C.-W.; Liu, L.-L.; Chou, Y.; Chang, W.-B.; Su, Y.-D.; et al. Towards the Small and the Beautiful: A Small Dibromotyrosine Derivative from Pseudoceratina sp. Sponge Exhibits Potent Apoptotic Effect through Targeting IKK/NFκB Signaling Pathway. Mar. Drugs 2013, 11, 3168–3185. [Google Scholar] [CrossRef] [PubMed]
- Shih, S.-P.; Lu, M.-C.; El-Shazly, M.; Lin, Y.-H.; Chen, C.-L.; Yu, S.S.-F.; Liu, Y.-C. The Antileukemic and Anti-Prostatic Effect of Aeroplysinin-1 Is Mediated through ROS-Induced Apoptosis via NOX Activation and Inhibition of HIF-1a Activity. Life 2022, 12, 687. [Google Scholar] [CrossRef] [PubMed]
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All Tangled up: How Cells Direct, Manage and Exploit Topoisomerase Function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ignazio, L.; Batie, M.; Rocha, S. Hypoxia and Inflammation in Cancer, Focus on HIF and NF-ΚB. Biomedicines 2017, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Nitulescu, G.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.; Grădinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt Pathway in Oncology Therapy and beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.-H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Stuhldreier, F.; Kassel, S.; Schumacher, L.; Wesselborg, S.; Proksch, P.; Fritz, G. Pleiotropic Effects of Spongean Alkaloids on Mechanisms of Cell Death, Cell Cycle Progression and DNA Damage Response (DDR) of Acute Myeloid Leukemia (AML) Cells. Cancer Lett. 2015, 361, 39–48. [Google Scholar] [CrossRef]
- Yun, C.W.; Kim, H.J.; Lim, J.H.; Lee, S.H. Heat Shock Proteins: Agents of Cancer Development and Therapeutic Targets in Anti-Cancer Therapy. Cells 2019, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Skok, Ž.; Zidar, N.; Kikelj, D.; Ilaš, J. Dual Inhibitors of Human DNA Topoisomerase II and Other Cancer-Related Targets. J. Med. Chem. 2020, 63, 884–904. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Kim, J.-H.; Kim, J.E.; Song, G.-Y.; Zhou, W.; Goh, S.-H.; Na, M.; Oh, S. Cytotoxic Activity of Aeroplysinin-1 against Colon Cancer Cells by Promoting β-Catenin Degradation. Food Chem. Toxicol. 2016, 93, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, M.-H.; Leake, R.E.; Rinaldi, F.; Müller-Klieser, W.; Maidhof, A.; Müller, W.E.G.; Schröder, H.C. Inhibition of Intrinsic Protein Tyrosine Kinase Activity of EGF-Receptor Kinase Complex from Human Breast Cancer Cells by the Marine Sponge Metabolite (+)-Aeroplysinin-1. Comp. Biochem. Physiol. B Biochem. 1990, 97, 151–158. [Google Scholar] [CrossRef]
- Venables, D.A.; Concepción, G.P.; Matsumoto, S.S.; Barrows, L.R.; Ireland, C.M. Makaluvamine N: A New Pyrroloiminoquinone from Zyzzya Fuliginosa. J. Nat. Prod. 1997, 60, 408–410. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.W.; Harper, M.K.; Faulkner, D.J. Makaluvamines H-M and Damirone C from the Pohnpeian Sponge Zyzzya Fuliginosa. J. Nat. Prod. 1995, 58, 1861–1867. [Google Scholar] [CrossRef]
- Carney, J.R.; Scheuer, P.J.; Kelly-Borges, M. Makaluvamine G, a Cytotoxic Pigment from an an Indonesian Sponge Histodermella sp. Tetrahedron 1993, 49, 8483–8486. [Google Scholar] [CrossRef]
- Radisky, D.C.; Radisky, E.S.; Barrows, L.R.; Copp, B.R.; Kramer, R.A.; Ireland, C.M. Novel Cytotoxic Topoisomerase II Inhibiting Pyrroloiminoquinones from Fijian Sponges of the Genus Zyzzya. J. Am. Chem. Soc. 1993, 115, 1632–1638. [Google Scholar] [CrossRef]
- Barrows, L.R.; Radisky, D.C.; Copp, B.R.; Swaffar, D.S.; Kramer, R.A.; Warters, R.L.; Ireland, C.M. Makaluvamines, Marine Natural Products, Are Active Anti-Cancer Agents and DNA Topo II Inhibitors. Anticancer Drug Des. 1993, 8, 333–347. [Google Scholar]
- Matsumoto, S.S.; Haughey, H.M.; Schmehl, D.M.; Venables, D.A.; Ireland, C.M.; Holden, J.A.; Barrows, L.R. Makaluvamines Vary in Ability to Induce Dose-Dependent DNA Cleavage via Topoisomerase II Interaction. Anticancer Drugs 1999, 10, 39–45. [Google Scholar] [CrossRef]
- Shinkre, B.A.; Raisch, K.P.; Fan, L.; Velu, S.E. Analogs of the Marine Alkaloid Makaluvamines: Synthesis, Topoisomerase II Inhibition, and Anticancer Activity. Bioorg. Med. Chem. Lett. 2007, 17, 2890–2893. [Google Scholar] [CrossRef] [Green Version]
- Boucle, S.; Melin, C.; Clastre, M.; Guillard, J. Design, Synthesis and Evaluation of New Marine Alkaloid-Derived Pentacyclic Structures with Anti-Tumoral Potency. Mar. Drugs 2015, 13, 655–665. [Google Scholar] [CrossRef] [Green Version]
- Guzmán, E.A.; Johnson, J.D.; Carrier, M.K.; Meyer, C.I.; Pitts, T.P.; Gunasekera, S.P.; Wright, A.E. Selective Cytotoxic Activity of the Marine-Derived Batzelline Compounds against Pancreatic Cancer Cell Lines. Anticancer Drugs 2009, 20, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Mizushina, Y. Molecular Action Mode of Hippospongic Acid A, an Inhibitor of Gastrulation of Starfish Embryos. J. Biochem. 2003, 133, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Su, J.-H.; Tseng, S.-W.; Lu, M.-C.; Liu, L.-L.; Chou, Y.; Sung, P.-J. Cytotoxic C21 and C22 Terpenoid-Derived Metabolites from the Sponge Ircinia sp. J. Nat. Prod. 2011, 74, 2005–2009. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.-C.; El-Shazly, M.; Juan, Y.-S.; Chang, C.-Y.; Su, J.-H.; Chen, Y.-C.; Shih, S.-P.; Chen, H.-M.; Wu, Y.-C.; Lu, M.-C. Cracking the Cytotoxicity Code: Apoptotic Induction of 10-Acetylirciformonin B Is Mediated through ROS Generation and Mitochondrial Dysfunction. Mar. Drugs 2014, 12, 3072–3090. [Google Scholar] [CrossRef] [Green Version]
- Su, J.-H.; Chang, W.-B.; Chen, H.-M.; El-Shazly, M.; Du, Y.-C.; Kung, T.-H.; Chen, Y.-C.; Sung, P.-J.; Ho, Y.-S.; Kuo, F.-W.; et al. 10-Acetylirciformonin B, a Sponge Furanoterpenoid, Induces DNA Damage and Apoptosis in Leukemia Cells. Molecules 2012, 17, 11839–11848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, D.J.; Miyamoto, S. Hexokinase II Integrates Energy Metabolism and Cellular Protection: Akting on Mitochondria and TORCing to Autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Okamoto, T.; Hayashi, K.; Yokoyama, N.; Sasaki, T.; Kitagawa, I. Marine Natural Products. XXXII. Absolute Configurations of C-4 of the Manoalide Family, Biologically Active Sesterterpenes from the Marine Sponge Hyrtios Erecta. Chem. Pharm. Bull. 1994, 42, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.-H.; Peng, B.-R.; Hsu, Y.-M.; El-Shazly, M.; Du, Y.-C.; Lu, M.-C.; Su, J.-H.; Liu, Y.-C. The Configuration-Dependent Anti-Leukemic Effect of Manoalide Stereoisomers: Reignite Research Interest in These Sponge-Derived Sesterterpenoids. Bioorg. Chem. 2021, 114, 105150. [Google Scholar] [CrossRef]
- Lee, M.-G.; Liu, Y.-C.; Lee, Y.-L.; El-Shazly, M.; Lai, K.-H.; Shih, S.-P.; Ke, S.-C.; Hong, M.-C.; Du, Y.-C.; Yang, J.-C.; et al. Heteronemin, a Marine Sesterterpenoid-Type Metabolite, Induces Apoptosis in Prostate LNcap Cells via Oxidative and ER Stress Combined with the Inhibition of Topoisomerase II and Hsp90. Mar. Drugs 2018, 16, E204. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-C.S.H.; Li, Z.-L.; Huang, T.-Y.; Su, K.-W.; Lin, C.-Y.; Huang, C.-H.; Chen, H.-Y.; Lu, M.-C.; Huang, H.-M.; Lee, S.-Y.; et al. Effect of Estrogen on Heteronemin-Induced Anti-Proliferative Effect in Breast Cancer Cells with Different Estrogen Receptor Status. Front. Cell Dev. Biol. 2021, 9, 688607. [Google Scholar] [CrossRef]
- Chung, C.-C.; Huang, T.-Y.; Chu, H.-R.; De Luca, R.; Candelotti, E.; Huang, C.-H.; Yang, Y.-C.S.H.; Incerpi, S.; Pedersen, J.Z.; Lin, C.-Y.; et al. Heteronemin and Tetrac Derivatives Suppress Non-Small Cell Lung Cancer Growth via ERK1/2 Inhibition. Food Chem. Toxicol. 2022, 161, 112850. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-T.; Bow, Y.-D.; Fu, P.-J.; Li, C.-Y.; Wu, C.-Y.; Chang, Y.-H.; Teng, Y.-N.; Li, R.-N.; Lu, M.-C.; Liu, Y.-C.; et al. A Marine Terpenoid, Heteronemin, Induces Both the Apoptosis and Ferroptosis of Hepatocellular Carcinoma Cells and Involves the ROS and MAPK Pathways. Oxid. Med. Cell Longev. 2021, 2021, 7689045. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Ding, W.; Ji, X.; Ao, X.; Liu, Y.; Yu, W.; Wang, J. Molecular Mechanisms of Ferroptosis and Its Role in Cancer Therapy. J. Cell Mol. Med. 2019, 23, 4900–4912. [Google Scholar] [CrossRef] [PubMed]
- Neophytou, C.M.; Trougakos, I.P.; Erin, N.; Papageorgis, P. Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance. Cancers 2021, 13, 4363. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Zhou, R.; Ma, Z. Autophagy—Cell Survival and Death. In Autophagy: Biology and Diseases; Qin, Z.-H., Ed.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; Volume 1206, pp. 667–696. ISBN 9789811506017. [Google Scholar]
- Wu, S.-Y.; Sung, P.-J.; Chang, Y.-L.; Pan, S.-L.; Teng, C.-M. Heteronemin, a Spongean Sesterterpene, Induces Cell Apoptosis and Autophagy in Human Renal Carcinoma Cells. BioMed Res. Int. 2015, 2015, 738241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, M.; Cerella, C.; Eifes, S.; Chateauvieux, S.; Morceau, F.; Jaspars, M.; Dicato, M.; Diederich, M. Heteronemin, a Spongean Sesterterpene, Inhibits TNFα-Induced NF-ΚB Activation through Proteasome Inhibition and Induces Apoptotic Cell Death. Biochem. Pharmacol. 2010, 79, 610–622. [Google Scholar] [CrossRef] [Green Version]
- Nunes-Xavier, C.E.; Mingo, J.; López, J.I.; Pulido, R. The Role of Protein Tyrosine Phosphatases in Prostate Cancer Biology. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 102–113. [Google Scholar] [CrossRef]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. ΓH2AX and Cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef]
- Saikia, M.; Retnakumari, A.P.; Anwar, S.; Anto, N.P.; Mittal, R.; Shah, S.; Pillai, K.S.; Balachandran, V.S.; Peter, V.; Thomas, R.; et al. Heteronemin, a Marine Natural Product, Sensitizes Acute Myeloid Leukemia Cells towards Cytarabine Chemotherapy by Regulating Farnesylation of Ras. Oncotarget 2018, 9, 18115–18127. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.-H.; Liu, Y.-C.; Su, J.-H.; El-Shazly, M.; Wu, C.-F.; Du, Y.-C.; Hsu, Y.-M.; Yang, J.-C.; Weng, M.-K.; Chou, C.-H.; et al. Antileukemic Scalarane Sesterterpenoids and Meroditerpenoid from Carteriospongia (Phyllospongia) sp., Induce Apoptosis via Dual Inhibitory Effects on Topoisomerase II and Hsp90. Sci. Rep. 2016, 6, 36170. [Google Scholar] [CrossRef]
- Shih, S.-P.; Lee, M.-G.; El-Shazly, M.; Juan, Y.-S.; Wen, Z.-H.; Du, Y.-C.; Su, J.-H.; Sung, P.-J.; Chen, Y.-C.; Yang, J.-C.; et al. Tackling the Cytotoxic Effect of a Marine Polycyclic Quinone-Type Metabolite: Halenaquinone Induces Molt 4 Cells Apoptosis via Oxidative Stress Combined with the Inhibition of HDAC and Topoisomerase Activities. Mar. Drugs 2015, 13, 3132–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.-C.; Lu, M.-C.; Hsu, K.-C.; El-Shazly, M.; Shih, S.-P.; Lien, S.-T.; Kuo, F.-W.; Yang, S.-C.; Chen, C.-L.; Yang, Y.-C.S.H. The Antileukemic Effect of Xestoquinone, a Marine-Derived Polycyclic Quinone-Type Metabolite, Is Mediated through ROS-Induced Inhibition of HSP-90. Molecules 2021, 26, 7037. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, G.P.; Foderaro, T.A.; Eldredge, G.S.; Lobkovsky, E.; Clardy, J.; Barrows, L.R.; Ireland, C.M. Topoisomerase II-Mediated DNA Cleavage by Adocia- and Xestoquinones from the Philippine Sponge Xestospongia sp. J. Med. Chem. 1995, 38, 4503–4507. [Google Scholar] [CrossRef]
- Evison, B.J.; Sleebs, B.E.; Watson, K.G.; Phillips, D.R.; Cutts, S.M. Mitoxantrone, More than Just Another Topoisomerase II Poison. Med. Res. Rev. 2016, 36, 248–299. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.R.; Scheuer, P.J.; Kelly-Borges, M. A New Bastadin from the Sponge (i) Psammaplysilla Purpurea (i). J. Nat. Prod. 1993, 56, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Juagdan, E.G.; Kalidindi, R.S.; Scheuer, P.J.; Kelly-Borges, M. Elenic Acid, an Inhibitor of Topoisomerase II, from a Sponge, Plakinastrella sp. Tetrahedron Lett. 1995, 36, 2905–2908. [Google Scholar] [CrossRef]
- Carney, J.R.; Scheuer, P.J. Popolohuanone E, a Topoisomerase-II Inhibitor with Selective Lung Tumor Cytotoxicity from the Pohnpei Sponge Dysidea sp. Tetrahedron Lett. 1993, 34, 3727–3730. [Google Scholar] [CrossRef]
- Yanagihara, M.; Sasaki-Takahashi, N.; Sugahara, T.; Yamamoto, S.; Shinomi, M.; Yamashita, I.; Hayashida, M.; Yamanoha, B.; Numata, A.; Yamori, T.; et al. Leptosins Isolated from Marine Fungus Leptoshaeria Species Inhibit DNA Topoisomerases I and/or II and Induce Apoptosis by Inactivation of Akt/Protein Kinase B. Cancer Sci. 2005, 96, 816–824. [Google Scholar] [CrossRef]
- Yamada, T.; Iritani, M.; Ohishi, H.; Tanaka, K.; Minoura, K.; Doi, M.; Numata, A. Pericosines, Antitumour Metabolites from the Sea Hare-Derived Fungus PEriconia Byssoides. Structures and Biological Activities. Org. Biomol. Chem. 2007, 5, 3979. [Google Scholar] [CrossRef]
- Usami, Y.; Ichikawa, H.; Arimoto, M. Synthetic Efforts for Stereo Structure Determination of Cytotoxic Marine Natural Product Pericosines as Metabolites of Periconia sp. from Sea Hare. Int. J. Mol. Sci. 2008, 9, 401–421. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, E52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Tian, X.; Huang, C.; Li, Q.; Zhang, S. Marinactinones A-C, New γ-Pyrones from Marine Actinomycete Marinactinospora Thermotolerans SCSIO 00606. J. Antibiot. 2011, 64, 189–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, Y.; Qi, X.; Li, D.; Zhu, T.; Mo, X. Anticancer Efficacy and Absorption, Distribution, Metabolism, and Toxicity Studies of Aspergiolide A in Early Drug Development. Drug Des. Dev. Ther. 2014, 2014, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhu, T.; Fang, Y.; Liu, H.; Gu, Q.; Zhu, W. Aspergiolide A, a Novel Anthraquinone Derivative with Naphtho[1,2,3-de]Chromene-2,7-Dione Skeleton Isolated from a Marine-Derived Fungus Aspergillus Glaucus. Tetrahedron 2007, 63, 1085–1088. [Google Scholar] [CrossRef]
- Ducroq, J.; Moha ou Maati, H.; Guilbot, S.; Dilly, S.; Laemmel, E.; Pons-Himbert, C.; Faivre, J.; Bois, P.; Stücker, O.; Le Grand, M. Dexrazoxane Protects the Heart from Acute Doxorubicin-Induced QT Prolongation: A Key Role for IKs. Br. J. Pharmacol. 2010, 159, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witchel, H.J. Drug-Induced HERG Block and Long QT Syndrome: HERG, Drugs, and LQTS. Cardiovasc. Ther. 2011, 29, 251–259. [Google Scholar] [CrossRef]
- Martinez-Farina, C.F.; McCormick, N.; Robertson, A.W.; Clement, H.; Jee, A.; Ampaw, A.; Chan, N.-L.; Syvitski, R.T.; Jakeman, D.L. Investigations into the Binding of Jadomycin DS to Human Topoisomerase IIβ by WaterLOGSY NMR Spectroscopy. Org. Biomol. Chem. 2015, 13, 10324–10327. [Google Scholar] [CrossRef]
- Raingeval, C.; Cala, O.; Brion, B.; Le Borgne, M.; Hubbard, R.E.; Krimm, I. 1D NMR WaterLOGSY as an Efficient Method for Fragment-Based Lead Discovery. J. Enzyme Inhib. Med. Chem. 2019, 34, 1218–1225. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Leung, I.K.H. Protein–Small Molecule Interactions by WaterLOGSY. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2019; Volume 615, pp. 477–500. ISBN 978-0-12-816762-5. [Google Scholar]
- Naine, S.J.; Devi, C.S.; Mohanasrinivasan, V.; Doss, C.G.P.; Kumar, D.T. Binding and Molecular Dynamic Studies of Sesquiterpenes (2R-Acetoxymethyl-1,3,3-Trimethyl-4t-(3-Methyl-2-Buten-1-Yl)-1t-Cyclohexanol) Derived from Marine Streptomyces sp. VITJS8 as Potential Anticancer Agent. Appl. Microbiol. Biotechnol. 2016, 100, 2869–2882. [Google Scholar] [CrossRef]
- Jemimah Naine, S.; Subathra Devi, C.; Mohanasrinivasan, V.; George Priya Doss, C. Bioactivity of Marine Streptomyces sp. VITJS4: Interactions of Cytotoxic Phthalate Derivatives with Human Topoisomerase II α: An in Silico Molecular Docking Analysis. Interdiscip. Sci. 2018, 10, 261–270. [Google Scholar] [CrossRef]
- El-Kashef, D.H.; Youssef, F.S.; Reimche, I.; Teusch, N.; Müller, W.E.G.; Lin, W.; Frank, M.; Liu, Z.; Proksch, P. Polyketides from the Marine-Derived Fungus Aspergillus Falconensis: In Silico and in Vitro Cytotoxicity Studies. Bioorg. Med. Chem. 2021, 29, 115883. [Google Scholar] [CrossRef]
- Wang, C.; Monger, A.; Wang, L.; Fu, P.; Piyachaturawat, P.; Chairoungdua, A.; Zhu, W. Precursor-Directed Generation of Indolocarbazoles with Topoisomerase IIα Inhibitory Activity. Mar. Drugs 2018, 16, E168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, C.; Numata, A.; Ito, Y.; Matsumura, E.; Araki, H.; Iwaki, H.; Kushida, K. Leptosins, Antitumour Metabolites of a Fungus Isolated from a Marine Alga. J. Chem. Soc. Perkin Trans. 1 1994, 13, 1859–1864. [Google Scholar] [CrossRef]
- Copp, B.R.; Ireland, C.M.; Barrows, L.R. Wakayin: A Novel Cytotoxic Pyrroloiminoquinone Alkaloid from the Ascidian Clavelina Species. J. Org. Chem. 1991, 56, 4596–4597. [Google Scholar] [CrossRef]
- Swaffar, D.S.; Ireland, C.M.; Barrows, L.R. A Rapid Mechanism-Based Screen to Detect Potential Anti-Cancer Agents. Anti-Cancer Drugs 1994, 5, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, I.; Bontemps, N.; Lahmy, S.; Banaigs, B.; Combaut, G.; Francisco, C.; Colson, P.; Houssier, C.; Waring, M.J.; Bailly, C. Binding to DNA and Cytotoxic Evaluation of Ascididemin, the Major Alkaloid from the Mediterranean Ascidian Cystodytes Dellechiajei. Anticancer Drug Des. 1995, 10, 333–346. [Google Scholar]
- Kobayash, J.; Cheng, J.; Nakamura, H.; Ohizumi, Y.; Hirata, Y.; Sasaki, T.; Ohta, T.; Nozoe, S. Ascididemin, a Novel Pentacyclic Aromatic Alkaloid with Potent Antileukemic Activity from the Okinawan Tunicate Didemnum sp. Tetrahedron Lett. 1988, 29, 1177–1180. [Google Scholar] [CrossRef]
- Lindsay, B.S.; Barrows, L.R.; Copp, B.R. Structural Requirements for Biological Activity of the Marine Alkaloid Ascididemin. Bioorg. Med. Chem. Lett. 1995, 5, 739–742. [Google Scholar] [CrossRef]
- Dassonneville, L.; Wattez, N.; Baldeyrou, B.; Mahieu, C.; Lansiaux, A.; Banaigs, B.; Bonnard, I.; Bailly, C. Inhibition of Topoisomerase II by the Marine Alkaloid Ascididemin and Induction of Apoptosis in Leukemia Cells. Biochem. Pharmacol. 2000, 60, 527–537. [Google Scholar] [CrossRef]
- Schmitz, F.J.; DeGuzman, F.S.; Choi, Y.-H.; Bilayet Hossain, B.; Rizvi, S.K.; van der Helm, D. Biologically Active Compounds from Marine Organisms. Pure Appl. Chem. 1990, 62, 1393–1396. [Google Scholar] [CrossRef]
- Matsumoto, S.S.; Biggs, J.; Copp, B.R.; Holden, J.A.; Barrows, L.R. Mechanism of Ascididemin-Induced Cytotoxicity. Chem. Res. Toxicol. 2003, 16, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Hasui, M.; Matsuda, M.; Yoshimatsu, S.; Okutani, K. Production of a Lactate-Associated Galactan Sulfate by a Dinoflagellate Gymnodinium A3. Fish. Sci. 1995, 61, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Umemura, K.; Yanase, K.; Suzuki, M.; Okutani, K.; Yamori, T.; Andoh, T. Inhibition of DNA Topoisomerases I and II, and Growth Inhibition of Human Cancer Cell Lines by a Marine Microalgal Polysaccharide. Biochem. Pharmacol. 2003, 66, 481–487. [Google Scholar] [CrossRef]
- Li, M.; Miao, Z.-H.; Chen, Z.; Chen, Q.; Gui, M.; Lin, L.-P.; Sun, P.; Yi, Y.-H.; Ding, J. Echinoside A, a New Marine-Derived Anticancer Saponin, Targets Topoisomerase2α by Unique Interference with Its DNA Binding and Catalytic Cycle. Ann. Oncol. 2010, 21, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Fortune, J.M.; Osheroff, N. Topoisomerase II as a Target for Anticancer Drugs: When Enzymes Stop Being Nice. Prog. Nucleic. Acid Res. Mol. Biol. 2000, 64, 221–253. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.J.; Corbett, A.H.; Osheroff, N. Effects of Topoisomerase II-Targeted Drugs on Enzyme-Mediated DNA Cleavage and ATP Hydrolysis: Evidence for Distinct Drug Interaction Domains on Topoisomerase II. Biochemistry 1993, 32, 3638–3643. [Google Scholar] [CrossRef]
- Liberio, M.S.; Sadowski, M.C.; Nelson, C.C.; Davis, R.A. Identification of Eusynstyelamide B as a Potent Cell Cycle Inhibitor Following the Generation and Screening of an Ascidian-Derived Extract Library Using a Real Time Cell Analyzer. Mar. Drugs 2014, 12, 5222–5239. [Google Scholar] [CrossRef] [Green Version]
- Liberio, M.S.; Sadowski, M.C.; Davis, R.A.; Rockstroh, A.; Vasireddy, R.; Lehman, M.L.; Nelson, C.C. The Ascidian Natural Product Eusynstyelamide B Is a Novel Topoisomerase II Poison That Induces DNA Damage and Growth Arrest in Prostate and Breast Cancer Cells. Oncotarget 2015, 6, 43944–43963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heck, M.M.; Earnshaw, W.C. Topoisomerase II: A Specific Marker for Cell Proliferation. J. Cell Biol. 1986, 103, 2569–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsay, B.S.; Christiansen, H.C.; Copp, B.R. Structural Studies of Cytotoxic Marine Alkaloids: Synthesis of Novel Ring-E Analogues of Ascididemin and Their in Vitro and in Vivo Biological Evaluation. Tetrahedron 2000, 56, 497–505. [Google Scholar] [CrossRef]
- Foderaro, T.A.; Barrows, L.R.; Lassota, P.; Ireland, C.M. Bengacarboline, a New β-Carboline from a Marine Ascidian Didemnum sp. J. Org. Chem. 1997, 62, 6064–6065. [Google Scholar] [CrossRef]
- Pouilhès, A.; Kouklovsky, C.; Langlois, Y.; Baltaze, J.-P.; Vispé, S.; Annereau, J.-P.; Barret, J.-M.; Kruczynski, A.; Bailly, C. Synthesis and Biological Evaluation of Bengacarboline Derivatives. Bioorg. Med. Chem. Lett. 2008, 18, 1212–1216. [Google Scholar] [CrossRef] [PubMed]
- Angel de la Fuente, J.; Jesús Martín, M.; del Mar Blanco, M.; Pascual-Alfonso, E.; Avendaño, C.; Carlos Menéndez, J. A C-Ring Regioisomer of the Marine Alkaloid Meridine Exhibits Selective in Vitro Cytotoxicity for Solid Tumours. Bioorg. Med. Chem. 2001, 9, 1807–1814. [Google Scholar] [CrossRef]
- Seo, Y.H. Dual Inhibitors Against Topoisomerases and Histone Deacetylases. J. Cancer Prev. 2015, 20, 85–91. [Google Scholar] [CrossRef]
- McClendon, A.K.; Osheroff, N. DNA Topoisomerase II, Genotoxicity, and Cancer. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2007, 623, 83–97. [Google Scholar] [CrossRef] [Green Version]
- Matias-Barrios, V.M.; Radaeva, M.; Song, Y.; Alperstein, Z.; Lee, A.R.; Schmitt, V.; Lee, J.; Ban, F.; Xie, N.; Qi, J.; et al. Discovery of New Catalytic Topoisomerase II Inhibitors for Anticancer Therapeutics. Front. Oncol 2021, 10, 633142. [Google Scholar] [CrossRef]

























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greco, G.; Pellicioni, V.; Cruz-Chamorro, I.; Attisani, G.; Stefanelli, C.; Fimognari, C. Marine-Derived Compounds Targeting Topoisomerase II in Cancer Cells: A Review. Mar. Drugs 2022, 20, 674. https://doi.org/10.3390/md20110674
Greco G, Pellicioni V, Cruz-Chamorro I, Attisani G, Stefanelli C, Fimognari C. Marine-Derived Compounds Targeting Topoisomerase II in Cancer Cells: A Review. Marine Drugs. 2022; 20(11):674. https://doi.org/10.3390/md20110674
Chicago/Turabian StyleGreco, Giulia, Valentina Pellicioni, Ivan Cruz-Chamorro, Giuseppe Attisani, Claudio Stefanelli, and Carmela Fimognari. 2022. "Marine-Derived Compounds Targeting Topoisomerase II in Cancer Cells: A Review" Marine Drugs 20, no. 11: 674. https://doi.org/10.3390/md20110674