
New Alkylpyridinium Anthraquinone, Isocoumarin, C-Glucosyl Resorcinol Derivative and Prenylated Pyranoxanthones from the Culture of a Marine Sponge-Associated Fungus, Aspergillus stellatus KUFA 2017
 , , , , , , and
, , , , , , and 
Abstract
:1. Introduction
2. Results and Discussion
3. Experimental Sections
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Extraction and Isolation
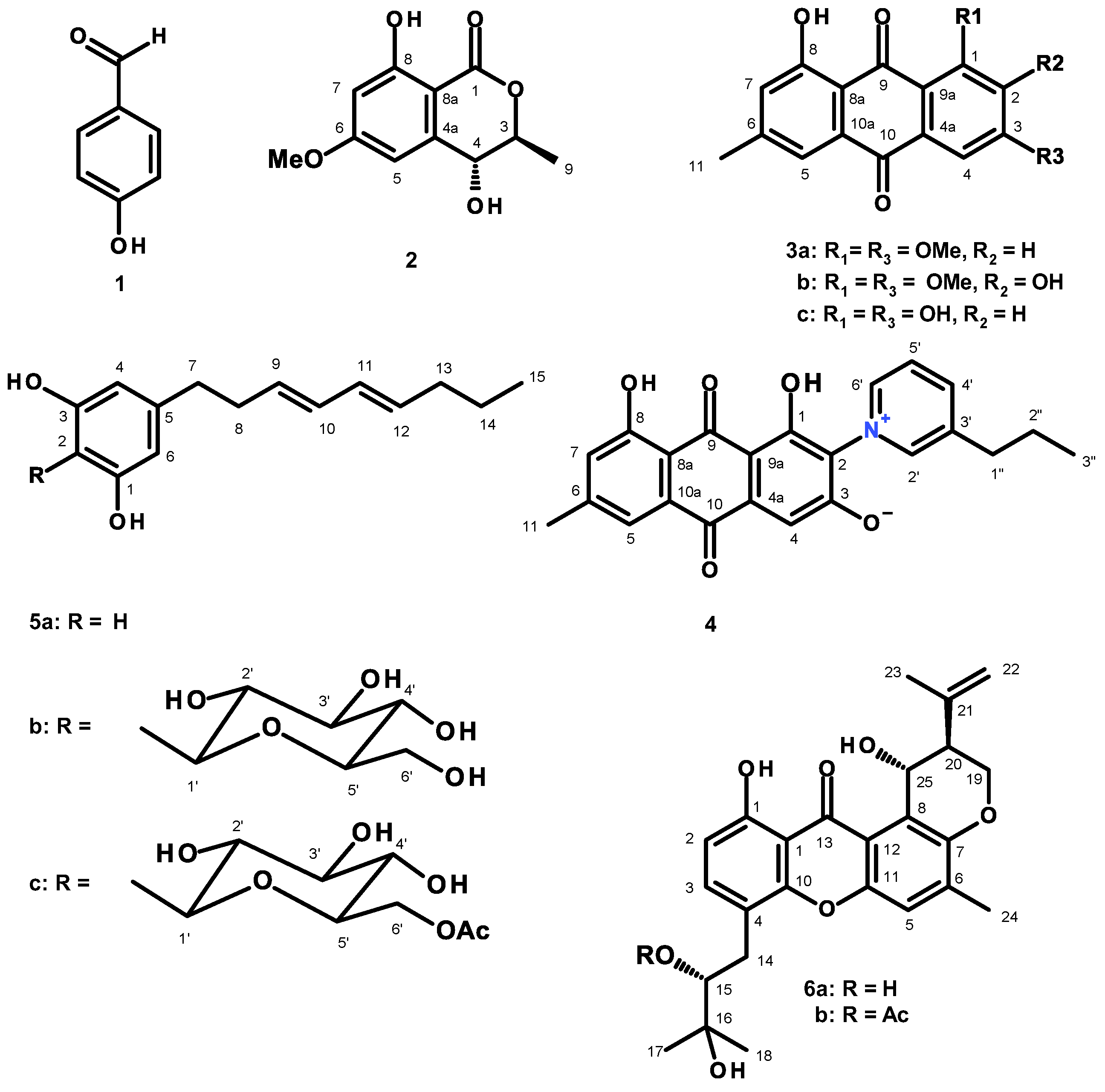
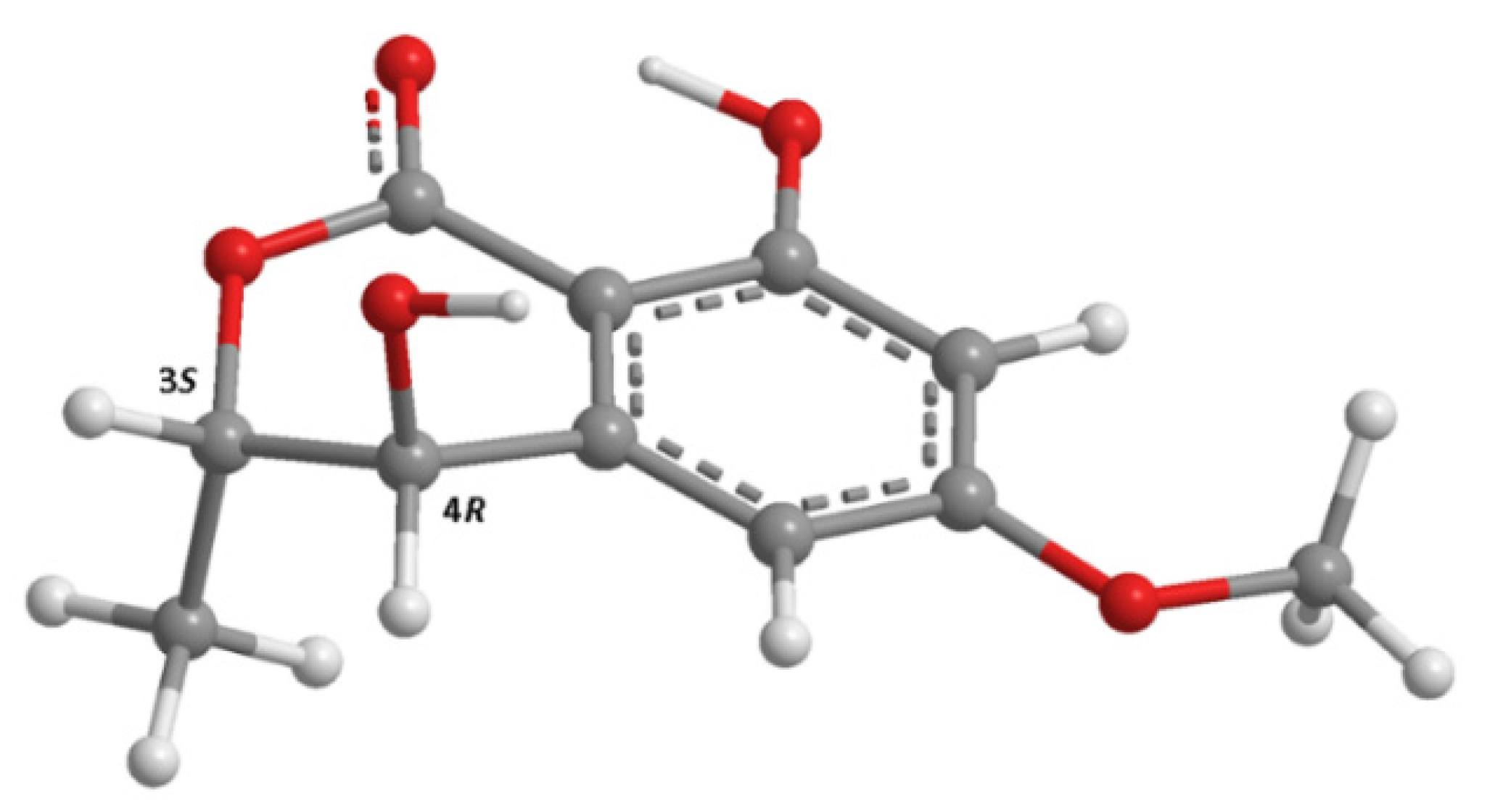
3.3.1. (3S,4R)-4-Hydroxy-6-Methoxymellein (2)
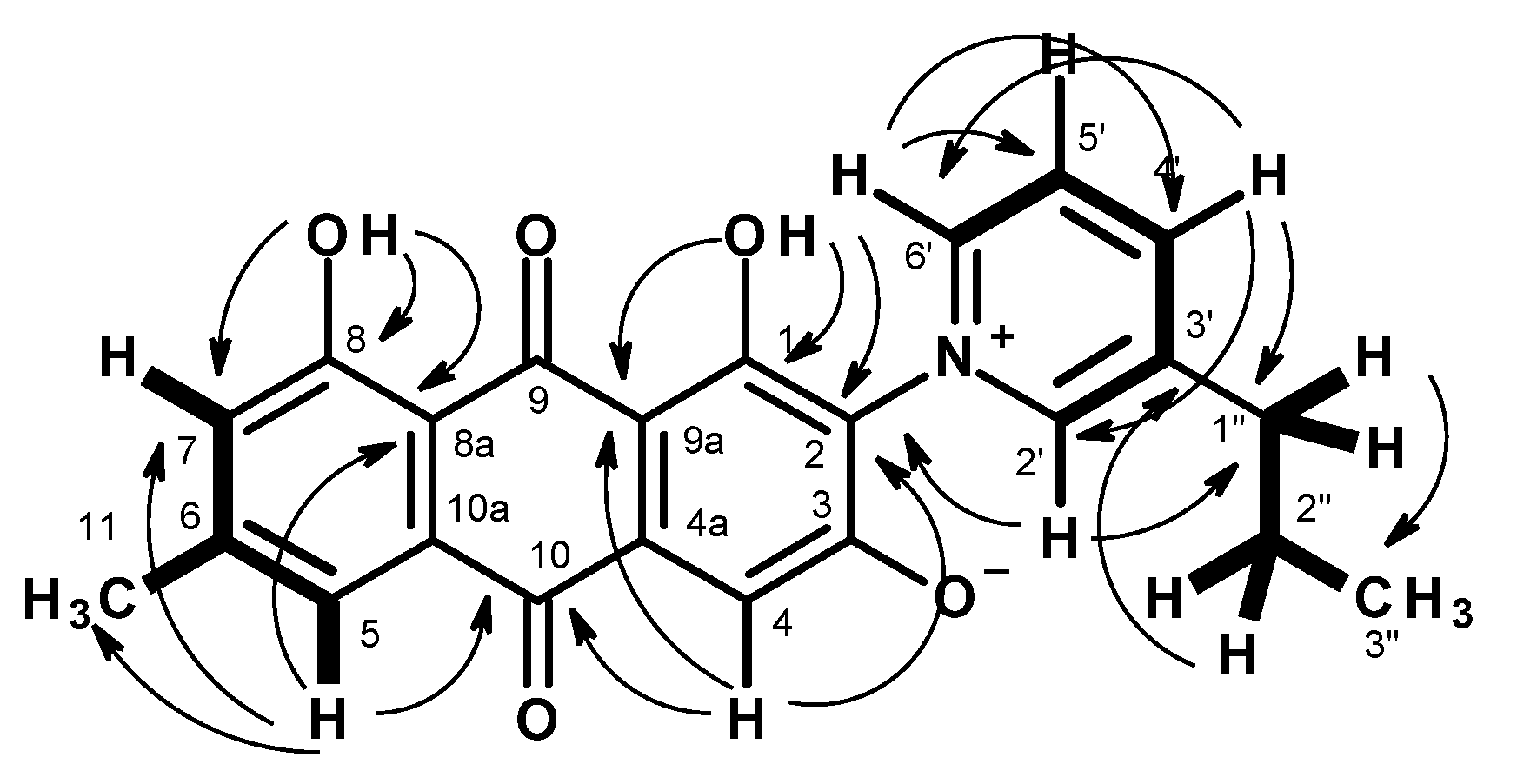
3.3.2. Stellatanthraquinone (4)
3.3.3. Carnemycin E (5b)
3.3.4. Acetyl Carnemycin E (5c)
3.4. X-ray Crystal Structures
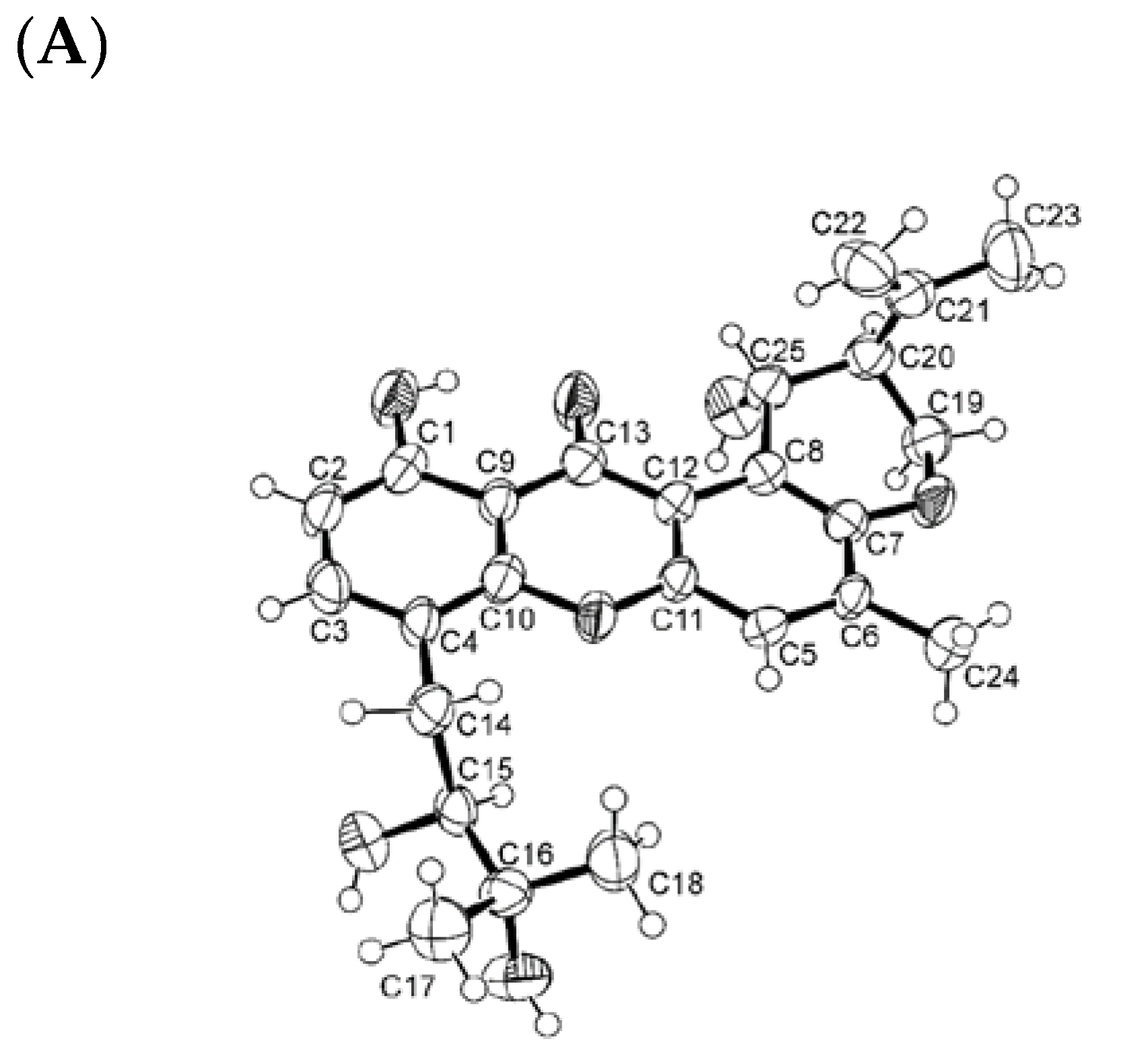
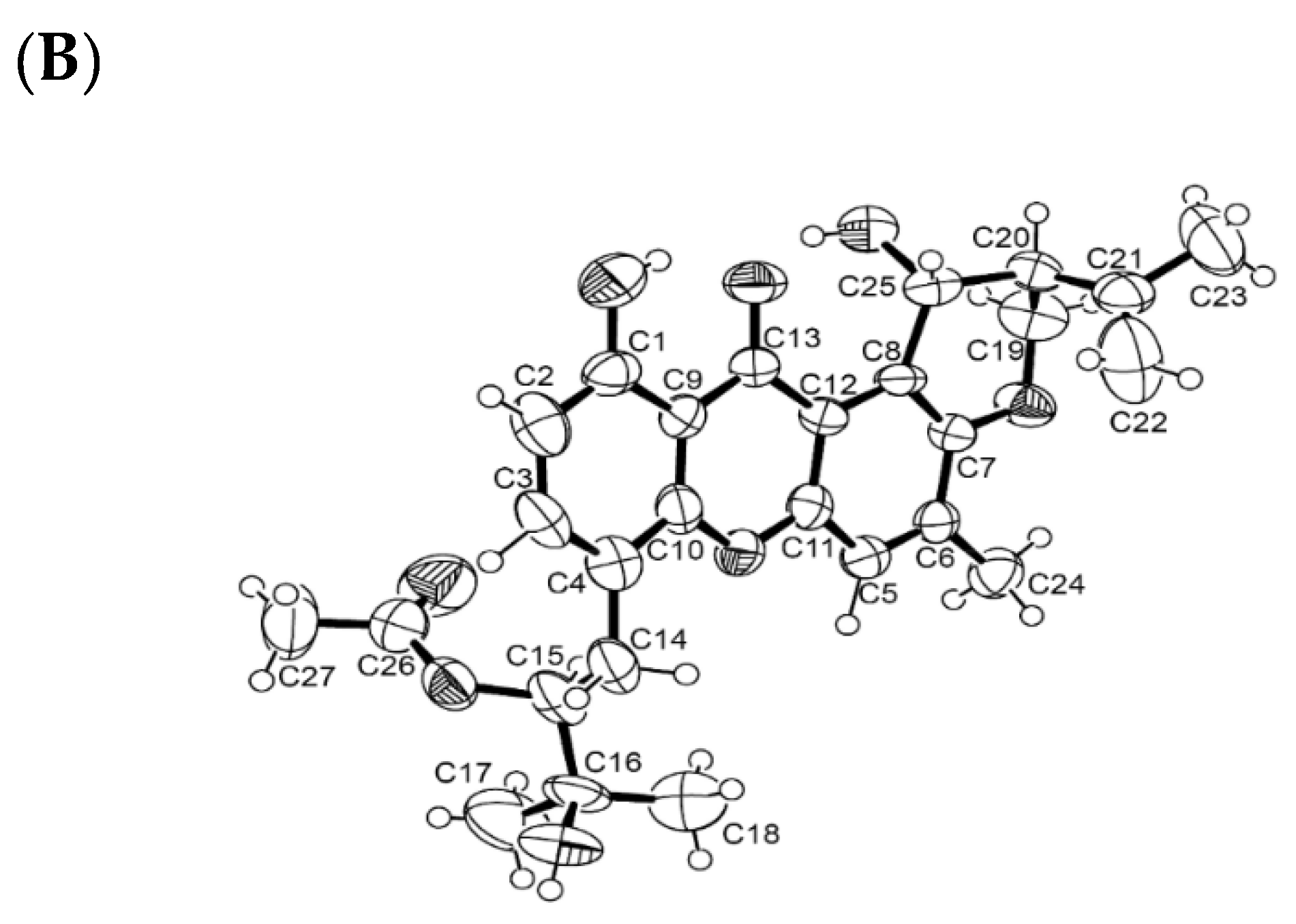
3.4.1. X-ray Crystal Structure of 6a
3.4.2. X-ray Crystal Structure of 6b
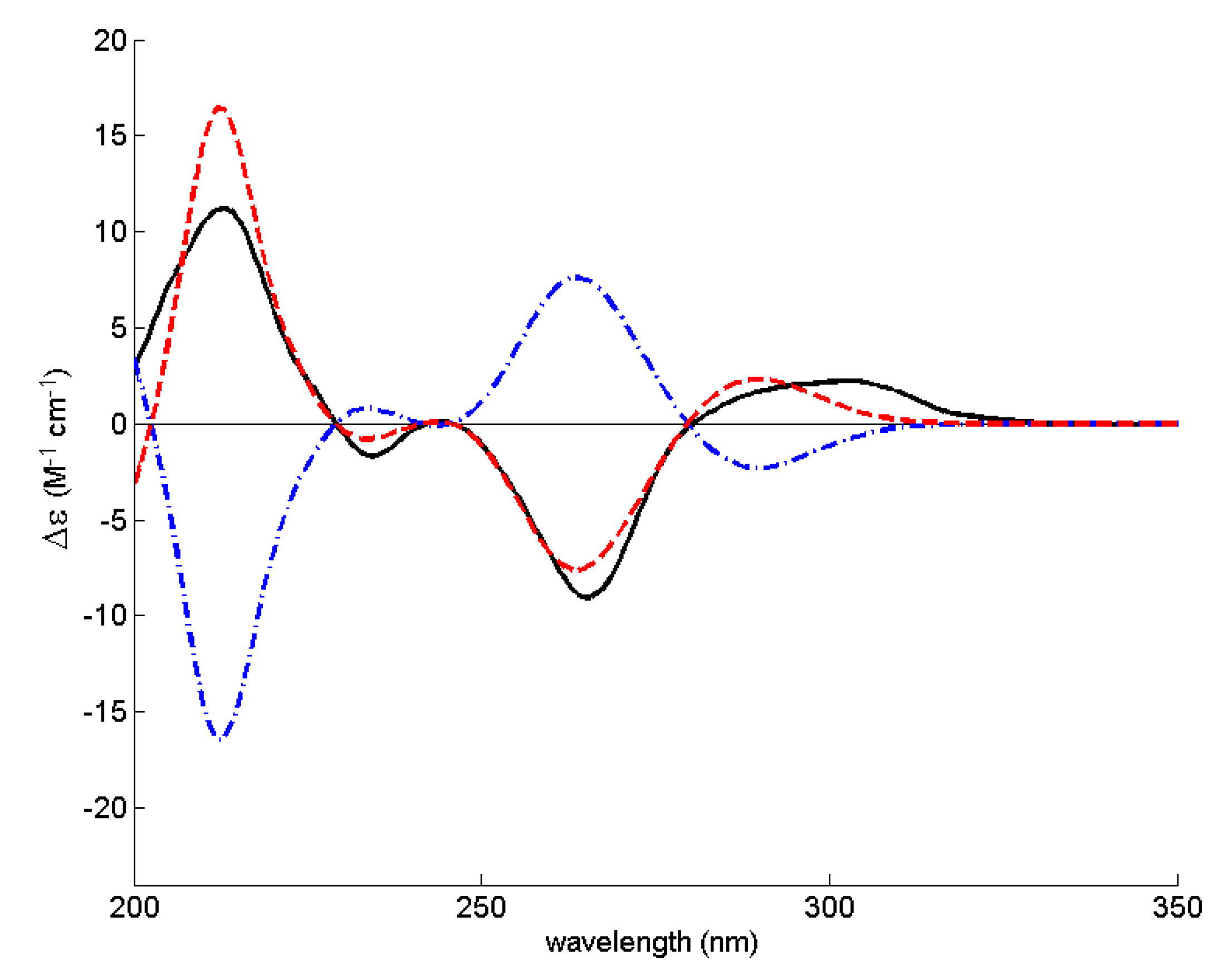
3.5. Electronic Circular Dichroism (ECD)
3.6. Antibacterial Activity Bioassays
3.6.1. Bacterial Strains and Testing Conditions
3.6.2. Antimicrobial Susceptibility Testing
3.6.3. Antibiotic Synergy Testing
3.6.4. Biofilm Formation Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.-C.; Zhuang, W.-Y. New species of Aspergillus (Aspergillaceae) from tropical islands of China. J. Fungi 2022, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- Hagestad, O.C.; Andersen, J.H.; Altermark, B.E.; Rämä, T. Cultivable marine fungi from the Arctic Archipelago of Svalbard and their antibacterial activity. Mycology 2020, 11, 230–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, F.P.; Kumla, D.; Pereira, J.A.; Sousa, E.; Dethoup, T.; Freitas-Silva, J.; Costa, P.M.; Mistry, S.; Silva, A.M.S.; Kijjoa, A. Prenylated phenylbutyrolactones from cultures of a marine sponge-associated fungus Aspergillus flavipes KUFA1152. Phytochemistry 2021, 185, 112709. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-T.; Xue, Y.-R.; Liu, C.-H. A brief review of bioactive metabolites derived from deep-sea fungi. Mar. Drugs 2015, 13, 4594–4616. [Google Scholar] [CrossRef] [Green Version]
- Samson, R.; Visagie, C.M.; Houbraken, J.; Hong, S.B.; Hubka, V.; Klaassen, C.H.W.; Perrone, G.; Seifert, K.A.; Susca, A.; Tanney, J.B.; et al. Phylogeny, identification and nomenclature of the genus Aspergillus. Stud. Mycol. 2014, 78, 141–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orfali, R.; Aboseada, M.A.; Abdel-Wahab, N.M.; Hassan, H.M.; Perveen, S.; Ameen, F.; Alturki, E.; Abdelmohsen, U.R. Recent updates on the bioactive compounds of the marine-derived genus Aspergillus. RSC Adv. 2021, 11, 17116–17150. [Google Scholar] [CrossRef]
- Kruger, G.J.; Steyn, P.S.; Vleggaar, R.; Rabie, C.J. X-ray crystal structure of astelotoxin, a novel mycotoxin from Aspergillus stellatus Curzi. J. Chem. Soc. Chem. Comm. 1979, 10, 441–442. [Google Scholar] [CrossRef]
- Kamal, A.; Husain, S.A.; Noorani, R.; Murtaza, N.; Qureshi, I.H.; Quershi, A.A. Studies in the biochemistry of microorganisms. XI. Isolation of tajixanthone, shamixanthone, ajamxanthone, shahenxanthone, najamxanthone, radixanthone and mannitol from mycelium of Aspergillus stellatus, Cruzi. J. Sci. Indus. Res. 1970, 13, 251–255. [Google Scholar]
- Prompanya, C.; Dethoup, T.; Bessa, L.J.; Pinto, M.M.M.; Gales, L.; Costa, P.M.; Silva, A.M.S.; Kijjoa, A. New isocoumarin derivatives and meroterpenoids from the marine sponge-associated fungus Aspergillus similanensis sp. nov. KUFA 0013. Mar. Drugs 2014, 12, 5160–5173. [Google Scholar] [CrossRef] [Green Version]
- Manojlović, I.; Bogdanović-Dusanović, G.; Gritsanapan, W.; Manojlović, N. Isolation and identification of anthraquinones of Caloplaca cerina and Cassia tora. Chem. Pap. 2006, 60, 466–468. [Google Scholar] [CrossRef]
- Hawas, U.W.; El-Beih, A.A.; El-Halawany, A.M. Bioactive anthraquinones from endophytic fungus Aspergillus versicolor isolated from red sea algae. Arch. Pharm. Res. 2012, 35, 1749–1756. [Google Scholar] [CrossRef] [PubMed]
- Noinart, J.; Buttachon, S.; Dethoup, T.; Gales, L.; Pereira, J.A.; Urbatzka, R.; Freitas, S.; Lee, M.; Silva, A.M.S.; Pinto, M.M.M.; et al. A new ergosterol analog, a new bis-anthraquinone and anti-obesity activity of anthraquinones from the marine sponge-associated fungus Talaromyces stipitatus KUFA 0207. Mar. Drugs 2017, 15, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.; Chen, C.; Sun, W.; Zang, Y.; Li, Q.; Wang, W.; Zeng, F.; Liu, J.; Zhou, Y.; Zhou, Q.; et al. Phenolic C-glycosides and aglycones from marine-derived Aspergillus sp. and their anti-inflammatory activities. J. Nat. Prod. 2019, 82, 1098–1106. [Google Scholar] [CrossRef]
- Pornpakakul, S.; Liangsakul, J.; Ngamrojanavanich, N.; Roengsumran, S.; Sihanonth, P.; Piapukiew, J.; Sangvichien, E.; Puthong, S.; Amorn Petsom, A. Cytotoxic activity of four xanthones from Emericella variecolor, an endophytic fungus isolated from Croton oblongifolius. Arch. Pharm. Res. 2006, 29, 140–144. [Google Scholar] [CrossRef]
- Figueroa, M.; González, M.C.; Rodríguez-Sotres, R.; Sosa-Peinado, A.; González-Andrade, M.; Cerda-Gárcıa-Rojas, C.M.; Mata, R. Calmodulin inhibitors from the fungus Emericella sp. Bioorg. Med. Chem. 2009, 17, 2167–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, A.M.; Rivera, C.J.; Sosa, P.A.; Figueroa, M.; Rodríguez-Sotres, R.; Mata, R. Development of the fluorescent biosensor h Calmodulin (h CaM) L39C-monobromobimane (mBBr)/V91C-mBBr, a novel tool for discovering new calmodulin inhibitors and detecting calcium. J. Med. Chem. 2011, 54, 3875–3884. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, Y.; Hamada, N.; Tanahashi, T. Aromatic compound from cultured lichen mycobionts of the three Graphis species. Heterocycles 2011, 83, 2157–2164. [Google Scholar]
- Damodaran, V.; Ryan, J.L.; Keyzers, R.A. Cyclic 3-alkyl pyridinium alkaloid monomers from a New Zealand Haliclona sp. marine sponge. J. Nat. Prod. 2013, 76, 1997–2001. [Google Scholar] [CrossRef]
- Schmidt, G.; Timm, C.; Köck, M.; Haliclocyclin, C. A new monomeric 3-alkyl pyridinium alkaloid from the arctic marine sponge Haliclona viscosa. Z. Naturforsch. 2011, 66b, 745–748. [Google Scholar] [CrossRef]
- Maarisit, W.; Abdjul, D.B.; Yamazaki, H.; Kato, H.; Rotinsulu, H.; Wewengkang, D.S.; Sumilat, D.A.; Kapojos, M.M.; Ukai, K.; Namikoshi, M. Anti-mycobacterial alkaloids, cyclic 3-alkyl pyridinium dimers, from the Indonesian marine sponge Haliclona sp. Bioorg. Med. Chem. Lett. 2017, 27, 3503–3506. [Google Scholar] [CrossRef]
- Markam, K.R.; Mues, R.; Stoll, M.; Zinsmeister, H.D. NMR spectra of flavone di-C-glycosides from Apometzgeria pubescens and the detection of rotational isomerism in 8-C-hexosylflavone. Z. Naturforsch. 1987, 42c, 1039–1042. [Google Scholar] [CrossRef]
- Chexal, K.K.; Fouweather, C.; Holker, J.S.E.; Simpson, T.J.; Young, K. The biosynthesis of fungal metabolites. Part III. Structure of shamixanthone and tajixanthone, metabolites of Aspergillus variecolor. J. Chem. Soc. Perkin Trans. 1974, 1, 1584–1593. [Google Scholar] [CrossRef]
- Chexal, K.K.; Holker, J.S.E.; Simpson, T.J. The biosynthesis of fungal metabolites. Part VI. Structure and biosynthesis of some minor metabolites from variant strains of Aspergillus variecolor. J. Chem. Soc. Perkin Trans. 1975, 1, 549–554. [Google Scholar] [CrossRef]
- Luís, A.; Cruz, C.; Duarte, A.P.; Domingues, F. An alkenylresorcinol derivative from Hakea sericea fruits and their antimicrobial activity. Nat. Prod. Com. 2013, 8, 1459–1462. [Google Scholar] [CrossRef] [Green Version]
- Hafez Ghoran, S.; Fatemeh Taktaz, F.; Ayatollahi, S.A.; Kijjoa, A. Anthraquinones and their analogues from marine-derived fungi: Chemistry and biological activities. Mar. Drugs 2022, 20, 474. [Google Scholar] [CrossRef] [PubMed]
- de Sá, J.D.M.; Pereira, J.A.; Dethoup, T.; Cidade, H.; Sousa, M.E.; Rodrigues, I.C.; Costa, P.M.; Mistry, S.; Silva, A.M.S.; Kijjoa, A. Anthraquinones, diphenyl ethers, and their derivatives from the culture of the marine sponge-associated fungus Neosartorya spinosa KUFA 1047. Mar. Drugs 2021, 19, 457. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef] [Green Version]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: New York, NY, USA, 1990; pp. 315–322. [Google Scholar]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 72, 5463–5467. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Flack, H.D. On enantiomorph-polarity estimation. Acta Crystallogr. Sect. A 1983, 39, 876–881. [Google Scholar] [CrossRef]
- Stephens, P.J.; Harada, N. ECD Cotton effect approximated by the Gaussian curve and other methods. Chirality 2010, 22, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Simões, R.R.; Aires-de-Sousa, M.; Conceição, T.; Antunes, F.; da Costa, P.M.; de Lencastre, H. High prevalence of EMRSA-15 in Portuguese public buses: A worrisome finding. PLoS ONE 2011, 6, e0017630. [Google Scholar] [CrossRef] [PubMed]
- Bessa, L.J.; Barbosa-Vasconcelos, A.; Mendes, Â.; Vaz-Pires, P.; Da Costa, P.M. High prevalence of multidrug-resistant Escherichia coli and Enterococcus spp. in river water, upstream and downstream of a wastewater treatment plant. J. Water Health 2014, 12, 426–435. [Google Scholar] [CrossRef]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing, 28th ed.; CLSI supplement M100; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2018. [Google Scholar]
- CLSI. Performance Standards for Antimicrobial Disk Susceptibility Tests; Approved Standard, 11th ed.; CLSI Document M02-A11; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 11th ed.; CLSI Standard M07; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2018. [Google Scholar]
- CLSI. Methods for Determining Bactericidal Activity of Antimicrobial Agents; Approved Guideline; CLSI Document M26-A; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 1999. [Google Scholar]
- Kumla, D.; Dethoup, T.; Gales, L.; Pereira, J.A.; Freitas-Silva, J.; Costa, P.M.; Silva, A.M.S.; Pinto, M.M.M.; Kijjoa, A. Erubescensoic acid, a new polyketide and a xanthonopyrone SPF-3059-26 from the culture of the marine sponge-associated fungus Penicillium erubescens KUFA 0220 and antibacterial activity evaluation of some of its constituents. Molecules 2019, 24, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, N.M.; Bessa, L.J.; Buttachon, S.; Costa, P.M.; Buaruang, J.; Dethoup, T.; Silva, A.M.S.; Kijjoa, A. Antibacterial and antibiofilm activities of tryptoquivalines and meroditerpenes isolated from the marine-derived fungi Neosartorya paulistensis, N. laciniosa, N. tsunodae, and the soil fungi N. fischeri and N. siamensis. Mar. Drugs 2014, 12, 822–839. [Google Scholar] [CrossRef] [Green Version]
- Odds, F.C. Synergy, antagonism, and what the chequer board puts between them. J. Antimicrob. Chemother. 2003, 52, 1. [Google Scholar] [CrossRef]
- Stepanović, S.; Vuković, D.; Hola, V.; Di Bonaventura, G.; Djuki’c, S.; Cirković, I.; Ruzicka, F. Quantification of biofilm in microtiter plates: Overview of testing conditions and practical recommendations for assessment of biofilm production by staphylococci. Apmis 2007, 115, 891–899. [Google Scholar] [CrossRef] [PubMed]



 ) and HMBC (
) and HMBC ( ) correlations in 4.
) correlations in 4.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δC, Type | δH, (J in Hz) | COSY | HMBC | NOESY |
|---|---|---|---|---|---|
| 1 | 169.1, CO | - | |||
| 3 | 77.8, CH | 4.63, dq (6.6, 2.0) | CH3-9 | CH3-9 | CH3-9 |
| 4 | 67.5, CH | 4.50, dd (5.6, 1.5) | OH-4 | C-4a, 5, 8a | H-5, CH3-9 |
| 4a | 142.1, C | - | - | ||
| 5 | 106.8, CH | 6.48, d (2.3) | H-7 | C-4, 6, 7, 8a | H-4, OCH3-6 |
| 6 | 166.2, C | - | |||
| 7 | 101.3, CH | 6.45, d (2.3) | H-5, | C-5, 6, 8a | OCH3-6 |
| 8 | 164.5, C | - | |||
| 8a | 100.0, C | - | |||
| 9 | 15.9, CH3 | 1.55, d (1.6) | H-3 | C-3, 4 | H-3, 4 |
| OCH3-6 | 55.8, CH3 | 3.85, s | C-6 | ||
| OH-4 | - | 2.28, m | C-3, 4 | ||
| OH-8 | - | 11.20, s | C-7, 8, 8a |
| Position | δC, Type | δH, (J in Hz) | COSY | HMBC |
|---|---|---|---|---|
| 1 | 159.5, C | |||
| 2 | 123.2, C | |||
| 3 | 171.9, C | |||
| 4 | 118.6, CH | 6.80, s | C-2, 9a, 10 | |
| 4a | 133.2, C | |||
| 5 | 120.4, CH | 7.46, d (1.0) | H-7 | C-7, 8a, 10, CH3-11 |
| 6 | 146.7, C | |||
| 7 | 124.4, CH | 7.11, s | H-5, CH3-11 | C-5, 8, 8a, CH3-11 |
| 8 | 161.2, C | |||
| 8a | 114.6, C | |||
| 9 | 184.5, CO | |||
| 9a | 100.0, C | |||
| 10 | 183.3, CO | |||
| 10a | 134.4, C | |||
| 11 | 21.9, CH3 | 2.40, s | H-7 | C-5, 6,7 |
| 2’ | 147.2, CH | 8.91, brs | H-4’ | C-2, 3’, 4’, 1” |
| 3’ | 142.8, C | |||
| 4’ | 146.2, CH | 8.57, d (8.0) | H-2’,5’ | C-2’, 6’, 1” |
| 5’ | 127.5, CH | 8.16, dd (8.0, 6.1) | H-4’, 6’ | C-3’, 6’ |
| 6’ | 145.6, CH | 8.85, d (6.1) | H-5’ | C-2, 4’, 5’ |
| 1” | 33.8, CH2 | 2.83, t (7.4) | H-2” | C-2’, 2”, 3’, 3” |
| 2” | 23.7, CH2 | 1.70, sex (7.4) | H-1”, 3” | C-1”, 3’, 3” |
| 3” | 13.7, CH3 | 0.94, t (7.3) | H-2” | C-1”, 3” |
| OH-1 | 13.44, s | C-1, 2, 9a | ||
| OH-8 | 12.50, s | C-7, 8, 8a |
| Position | δC, Type | δH, (J in Hz) | COSY | HMBC |
|---|---|---|---|---|
| 1 | 157.1, C | |||
| 2 | 110.2, C | |||
| 3 | 157.1, C | |||
| 4 | 107.6, CH | 6.11, s | C-1’, 3, 6, 7 | |
| 5 | 142.5, C | |||
| 6 | 107.6,CH | 6.11, s | C-1’, 2, 4, 7 | |
| 7 | 35.5, CH2 | 2.45, t (6.8) | H-8 | C-4, 5, 6, 8, 9 |
| 8 | 34.0, CH2 | 2.27, dd (14.3, 6.6) | H-7, H-9 | C-5, 7, 9, 10 |
| 9 | 131.8, CH | 5.62, m | H-8, 10 | C-10, 11 |
| 10 | 130.9, CH | 5.91, m | H-9, 11 | C-8, 9, 12 |
| 11 | 131.1, CH | 6.04, m | H-10, 12 | C-9, 13 |
| 12 | 132.6, CH | 5.55, m | H-11, 13 | C-10, 13, 14 |
| 13 | 34.5, CH2 | 2.00, dd (14.3, 7.2) | H-12, 14 | C-11, 12, 14, 15 |
| 14 | 22.5, CH2 | 1.36, sext (7.2) | H-13, 15 | C-12, 13, 15 |
| 15 | 14.0, CH3 | 0.87, t (7.2) | H-14 | C-13, 14 |
| 1’ | 75.0, CH | 4.62, d (9.6) | H-2’ | C-1, 2, 2’, 3, 3’ |
| 2’ | 72.1, CH | 3.74, m | ||
| 3’ | 79.1, CH | 3.22, m | ||
| 4’ | 70.3, CH | 3.22, m | OH-4’ | |
| 5’ | 81.5, CH | 3.20, m | ||
| 6’a b | 61.2, CH2 | 3.50, dd (11.0, 5.5) 3.65, dd (11.0, 5.2) | H-5’, 6’b, OH-6’ H-5’, 6’a, OH-6’ | |
| OH-3 | 8.67, s | C-2, 3, 4 | ||
| OH-4’ | 4.90, dd (10.7, 2.9) | H-4’ | C-4’, 5’ | |
| OH-6’ | 4.59, d (5.5) | H2-6’ | C-6’ |
| Position | δC, Type | δH, (J in Hz) | COSY | HMBC |
|---|---|---|---|---|
| 1 | 157.3, C | |||
| 2 | 109.9, C | |||
| 3 | 157.3, C | |||
| 4 | 107.5, CH | 6.11, s | C-1’, 2, 3, 6, 7 | |
| 5 | 142.5, C | |||
| 6 | 107.5, CH | 6.11, s | C-1’, 2, 3, 4, 7 | |
| 7 | 35.5, CH2 | 2.44, t (7.2) | H-8 | C-4, 5, 6, 8, 9 |
| 8 | 34.0, CH2 | 2.26, dd (14.7, 7.2) | H-7, 9 | C-7, 9, 10 |
| 9 | 131.8, CH | 5.59, ddd (14.6, 7.2, 7.2) | H-8, 10 | C-11 |
| 10 | 130.9, CH | 5.97, m | H-9, 11 | |
| 11 | 131.1, CH | 6.04, m | H-10, 12 | C-9 (w), 13 |
| 12 | 132.6, CH | 5.57, ddd (14.5, 7.7, 7.1) | H-11, 13 | C-10, 13, 14 |
| 13 | 34.5, CH2 | 2.01, m | H-12, 14 | C-11, 12, 14, 15 |
| 14 | 22.5, CH2 | 1.36, sex (7.4) | H-13, 15 | 12, 13, 15 |
| 15 | 14.0, CH3 | 0.86, t (7.4) | H-14 | C-13, 14 |
| 1’ | 74.9, CH | 4.60, d (9.8) | H-2’ | C-1, 2, 2’ 3, 3’ |
| 2’ | 71.5, CH | 3.83, t (9.2) | H-1’, 3’ | C-1’, 3’ |
| 3’ | 79.0, CH | 3.21, t (8.7) | H-4’ | C-4’ |
| 4’ | 70.6, CH | 3.18, t (8.7) | H-3’ | C-3’ |
| 5’ | 78.4, CH | 3.36 (under water peak) | H-6’b | C-4’ (w) |
| 6’a b | 64.8, CH2 | 4.32, d (11.6) 3.98 dd (11.6, 3.9) | H-6’b H-5’, 6’a | C-4’, CO (OAc) C-5’, 6’ |
| OAc | 170.9, CO | - | ||
| OAc | 21.2, CH3 | 2.00, s | C-6’ | |
| OH-3 | 8.74, brs | |||
| OH-3’ | 4.95, br | |||
| OH-4’ | 5.15, br |
| Compound | E. faecalis ATCC 29212 | E. faecalis B3/101 (VRE) | S. aureus ATCC 29213 | S. aureus 74/24 (MRSA) | ||||
|---|---|---|---|---|---|---|---|---|
| MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | |
| 2 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 3a | >32 | >32 | >32 | >32 | >32 | >32 | >32 | >32 |
| 3b | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 4 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | >32 |
| 5a | 16 | 32 | 16 | 64 | 32 | 32 | 16 | 32 |
| 5b | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 5c | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 6a | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 6b | >32 | >32 | >32 | >32 | >32 | >32 | >32 | >32 |
| VAN | 4 | - | - | - | - | - | - | - |
| OXA | - | - | - | - | 0.2 | - | - | - |
| Compound | Concentration | Biofilm Biomass (% of Control) | |
|---|---|---|---|
| E. faecalis ATCC 29212 | S. aureus ATCC 29213 | ||
| 5a | 64 µg/mL | - | 0.00 ± 0.06 *** |
| 32 µg/mL | 0.01 ± 0.01 *** | 0.04 ± 0.12 *** | |
| 16 µg/mL | 0.02 ± 0.01 *** | - | |
| DMSO | 1% (v/v) | 1.00 ± 0.03 *** | 1.00 ± 0.01 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado, F.P.; Rodrigues, I.C.; Gales, L.; Pereira, J.A.; Costa, P.M.; Dethoup, T.; Mistry, S.; Silva, A.M.S.; Vasconcelos, V.; Kijjoa, A. New Alkylpyridinium Anthraquinone, Isocoumarin, C-Glucosyl Resorcinol Derivative and Prenylated Pyranoxanthones from the Culture of a Marine Sponge-Associated Fungus, Aspergillus stellatus KUFA 2017. Mar. Drugs 2022, 20, 672. https://doi.org/10.3390/md20110672
Machado FP, Rodrigues IC, Gales L, Pereira JA, Costa PM, Dethoup T, Mistry S, Silva AMS, Vasconcelos V, Kijjoa A. New Alkylpyridinium Anthraquinone, Isocoumarin, C-Glucosyl Resorcinol Derivative and Prenylated Pyranoxanthones from the Culture of a Marine Sponge-Associated Fungus, Aspergillus stellatus KUFA 2017. Marine Drugs. 2022; 20(11):672. https://doi.org/10.3390/md20110672
Chicago/Turabian StyleMachado, Fátima P., Inês C. Rodrigues, Luís Gales, José A. Pereira, Paulo M. Costa, Tida Dethoup, Sharad Mistry, Artur M. S. Silva, Vitor Vasconcelos, and Anake Kijjoa. 2022. "New Alkylpyridinium Anthraquinone, Isocoumarin, C-Glucosyl Resorcinol Derivative and Prenylated Pyranoxanthones from the Culture of a Marine Sponge-Associated Fungus, Aspergillus stellatus KUFA 2017" Marine Drugs 20, no. 11: 672. https://doi.org/10.3390/md20110672