
Seven New Alkaloids Isolated from Marine Flavobacterium Tenacibaculum discolor sv11

Abstract
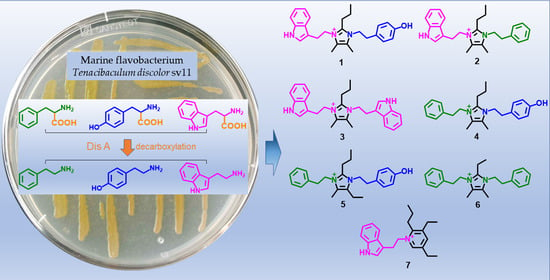
:
1. Introduction
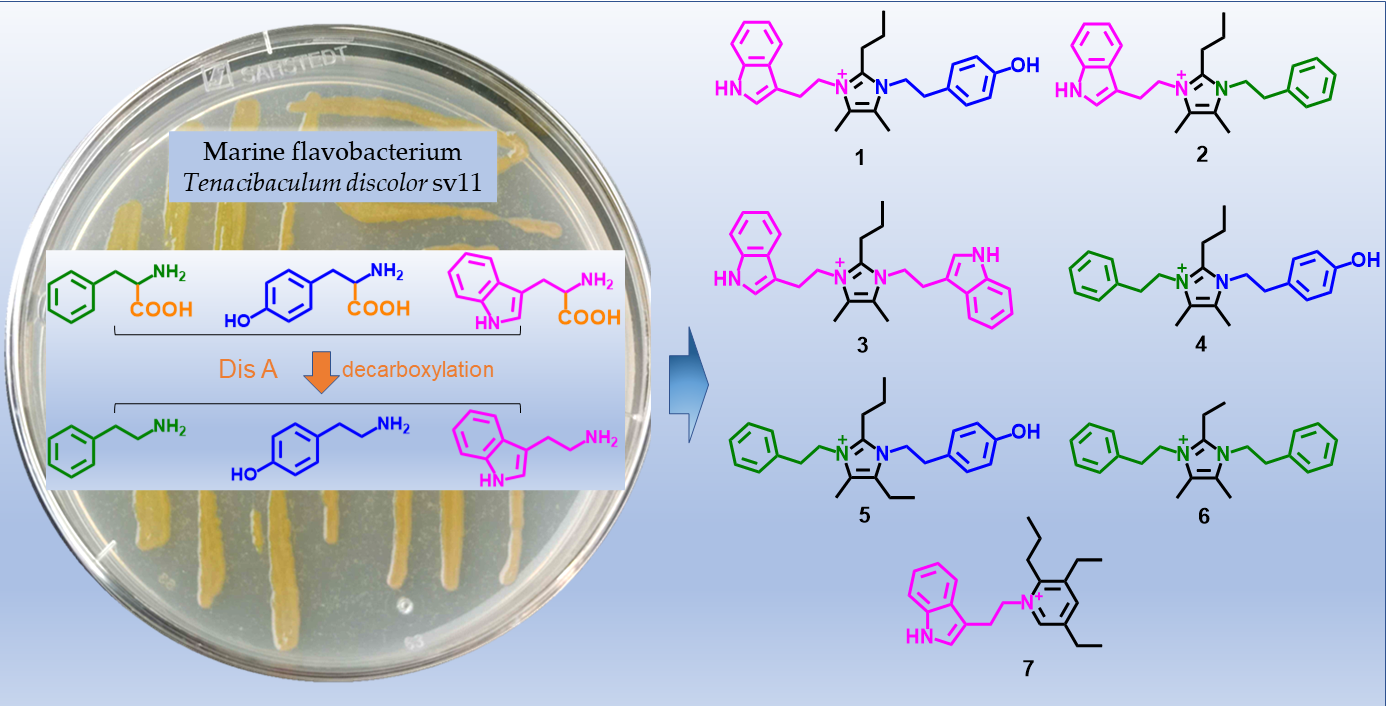
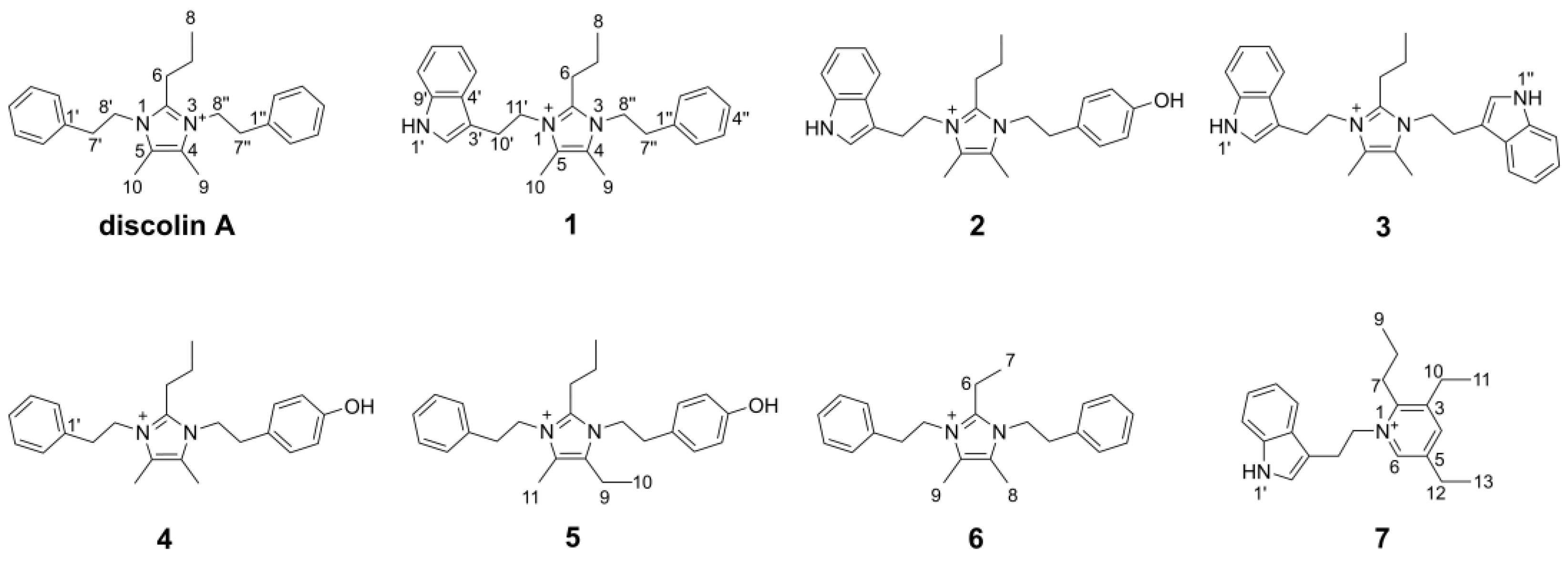
2. Results
3. Conclusions and Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Extraction and Isolation
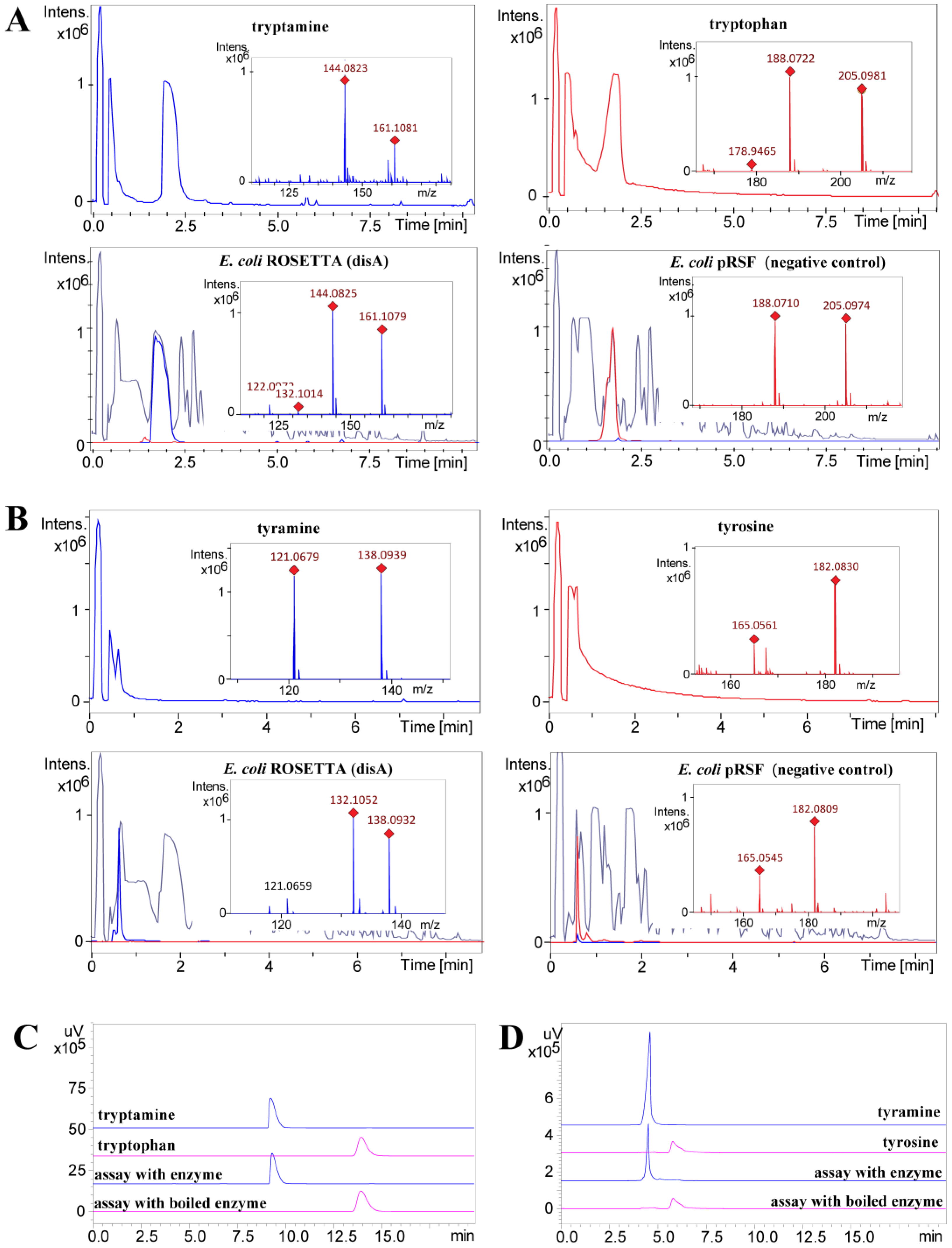
4.3. Enzymatic Activity of Dis A
4.4. Bioactivity Tests
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seipp, K.; Geske, L.; Opatz, T. Marine Pyrrole Alkaloids. Mar. Drugs 2021, 19, 514. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z. Muscarine, imidazole, oxazole and thiazole alkaloids. Nat. Prod. Rep. 2016, 33, 1268–1317. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lv, S.; Liu, J.; Yu, Y.; Wang, H.; Zhang, H. An Overview of Bioactive 1, 3-Oxazole-Containing Alkaloids from Marine Organisms. Pharmaceuticals 2021, 14, 1274. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D. Pyrrole, pyrrolidine, pyridine, piperidine and tropane alkaloids. Nat. Prod. Rep. 2000, 17, 435–446. [Google Scholar] [CrossRef]
- Ma, X.; Liang, X.; Huang, Z.H.; Qi, S.H. New alkaloids and isocoumarins from the marine gorgonian-derived fungus Aspergillus sp. SCSIO 41501. Nat. Prod. Res. 2000, 34, 1992–2000. [Google Scholar] [CrossRef]
- Hassan, W.; Edrada, R.; Ebel, R.; Wray, V.; Berg, A.; Soest, R.V.; Wiryowidagdo, S.; Proksch, P. New imidazole alkaloids from the Indonesian sponge Leucetta chagosensis. J. Nat. Prod. 2004, 67, 817–822. [Google Scholar] [CrossRef]
- Dyson, L.; Wright, A.D.; Young, K.A.; Sakoff, J.A.; McCluskey, A. Synthesis and anticancer activity of focused compound libraries form the natural product lead, oroidin. Bioorg. Med. Chem. 2014, 22, 1690–1699. [Google Scholar] [CrossRef]
- Liu, L.P.; Zong, M.H.; Linhardt, R.J.; Lou, W.Y.; Li, N.; Huang, C.; Wu, H. Mechanistic insights into the effect of imidazolium ionic liquid on liquid production by Geotrichum fermentas. Biotechnol. Biofuels 2016, 9, 266. [Google Scholar] [CrossRef]
- Johnson, N.A.; Southerland, M.R.; Youngs, W.J. Recent Developments in the Medicinal Applications of Silver-NHC Complexes and Imidazolium Salts. Molecules 2017, 22, 1263. [Google Scholar] [CrossRef]
- Kirchhecker, S.; Antonietti, M.; Esposito, D. Hydrothermal decarboxylation of amino acid derived imidazolium zwitterions: A sustainable approach towards ionic liquids. Green Chem. 2014, 16, 3705–3709. [Google Scholar] [CrossRef] [Green Version]
- Roué, M.; Domart-Coulon, I.; Ereskovsky, A.; Djediat, C.; Perez, T.; Bourguet-Kondracki, M.L. Cellular localization of clathridimine, an antimicrobial 2-aminoimidazole alkaloid produced by the Mediterranean calcareous sponge Clathrina clathrus. J. Nat. Prod. 2010, 73, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Bjørsvik, H.; Sandtorv, A. Synthesis of Imidazole Alkaloids Originated in Marine Sponges. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2014; Volume 42, pp. 33–57. [Google Scholar]
- Dunbar, D.C.; Rimoldi, J.M.; Clark, A.M.; Kelly, M.; Hamann, M.T. Anti-cryptococcal and nitric oxide synthase inhibitory imidazole alkaloids from the calcareous sponge Leucetta cf chagosensis. Tetrahedron 2000, 56, 8795–8798. [Google Scholar] [CrossRef]
- Gross, H.; Kehraus, S.; König, G.M.; Woerheide, G.; Wright, A.D. New and biologically active imidazole alkaloids from two sponges of the genus Leucetta. J. Nat. Prod. 2002, 65, 1190–1193. [Google Scholar] [CrossRef] [PubMed]
- Bernardet, J.F. Family I. Flavobacteriaceae Reichenbach 1992. In Bergey’s Manual of Systematic Bacteriology, 2nd ed.; Krieg, N.R., Staley, J.T., Brown, D.R., Hedlund, B.P., Paster, B.J., Ward, N.L., Ludwig, W., Whitman, W.B., Eds.; Springer: New York, NY, USA, 2011; Volume 4, pp. 106–111. [Google Scholar]
- Frette, L.; Jørgensen, N.O.; Irming, H.; Kroer, N. Tenacibaculum skagerrakense sp. nov., a marine bacterium isolated from the pelagic zone in Skagerrak, Denmark. Int. J. Syst. Evol. Microbiol. 2004, 54, 519–524. [Google Scholar] [CrossRef]
- Yoon, J.H.; Kang, S.J.; Jung, S.Y.; Oh, H.W.; Oh, T.K. Gaetbulimicrobium brevivitae gen. nov., sp. nov., a novel member of the family Flavobacteriaceae isolated from a tidal flat of the Yellow Sea in Korea. Int. J. Syst. Evol. Microbiol. 2006, 56, 115–119. [Google Scholar] [CrossRef]
- Heindl, H.; Wiese, J.; Imhoff, J.F. Tenacibaculum adriaticum sp. nov., from a bryozoan in the Adriatic Sea. Int. J. Syst. Evol. Microbiol. 2008, 58, 542–547. [Google Scholar] [CrossRef]
- Wang, J.T.; Chou, Y.J.; Chou, J.H.; Chen, C.A.; Chen, W.M. Tenacibaculum aiptasiae sp. nov., isolated from a sea anemone Aiptasia pulchella. Int. J. Syst. Evol. Microbiol. 2008, 58, 761–766. [Google Scholar] [CrossRef]
- Lee, Y.S.; Baik, K.S.; Park, S.Y.; Kim, E.M.; Lee, D.H.; Kahng, H.Y.; Jeon, C.O.; Jung, J.S. Tenacibaculum crassostreae sp. nov., isolated from the Pacific oyster, Crassostrea gigas. Int. J. Syst. Evol. Microbiol. 2009, 59, 1609–1614. [Google Scholar] [CrossRef]
- Pineiro-Vidal, M.; Riaza, A.; Santos, Y. Tenacibaculum discolor sp. nov. and Tenacibaculum gallaicum sp. nov., isolated from sole (Solea senegalensis) and turbot (Psetta maxima) culture systems. Int. J. Syst. Evol. Microbiol. 2008, 58, 21–25. [Google Scholar] [CrossRef]
- Suzuki, M.; Nakagawa, Y.; Harayama, S.; Yamamoto, S. Phylogenetic analysis and taxonomic study of marine Cytophaga-like bacteria: Proposal for Tenacibaculum gen. nov. with Tenacibaculum maritimum comb. nov. and Tenacibaculum ovolyticum comb. nov., and description of Tenacibaculum mesophilum sp. nov. and Tenacibaculum amylolyticum sp. nov. Int. J. Syst. Evol. Microbiol. 2001, 51, 1639–1652. [Google Scholar]
- Avendaño-Herrera, R.; Toranzo, A.E.; Magariños, B. Tenacibaculosis infection in marine fish caused by Tenacibaculum maritimum: A review. Dis. Aquat. Organ. 2006, 71, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y.; Ge, Y.; Zhou, T.; Sharma, A.R.; Harunari, E.; Oku, N.; Trianto, A. Tenacibactins K–M, cytotoxic siderophores from a coral-associated gliding bacterium of the genus Tenacibaculum. Beilstein. J. Org. Chem. 2022, 18, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Kanoh, K.; Adachi, K.; Matsuda, S.; Shizuri, Y. Tenacibactins a-d, hydroxamate siderophores from a marine-derived bacterium, Tenacibaculum sp. a4k-17. J. Nat. Prod. 2007, 70, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.J.; Nakano, K.; Sakai, R. Bisucaberin B, a Linear Hydroxamate Class Siderophore from the Marine Bacterium Tenacibaculum mesophilum. Molecules 2013, 18, 3917–3926. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Linares-Otoya, V.; Liu, Y.; Mettal, U.; Marner, M.; Armas Mantilla, L.; Willbold, S.; Kurtán, T.; Linares-Otoya, L.; Schäberle, T.F. Discovery and Biosynthesis of Antimicrobial Phenethylamine Alkaloids from the Marine Flavobacterium Tenacibaculum discolor sv11. J. Nat. Prod. 2022, 85, 1039–1051. [Google Scholar] [CrossRef]
- Yan, J.X.; Wu, Q.; Helfrich, E.J.N.; Chevrette, M.G.; Braun, D.R.; Heyman, H.; Ananiev, G.E.; Rajski, S.R.; Currie, C.R.; Clardy, J.; et al. Bacillimidazoles A-F, Imidazolium-Containing Compounds Isolated from a Marine Bacillus. Mar. Drugs 2022, 20, 43. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D.J.; Bryce, D.L. Spectrometric Identification of Organic Compounds, 8th ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2015; pp. 299–305. [Google Scholar]
- Zeng, Z.; Qasem, A.M.A.; Woodman, T.J.; Rowan, M.G.; Blagbrough, I.S. Impacts of Steric Compression, Protonation, and Intramolecular Hydrogen Bonding on the 15N NMR Spectroscopy of Norditerpenoid Alkaloids and Their Piperidine-Ring Analogues. ACS Omega 2020, 5, 14116–14122. [Google Scholar] [CrossRef]
- Wang, F.P.; Chen, D.L.; Deng, H.Y.; Chen, Q.H.; Liu, X.Y.; Jian, X.X. Further revisions on the diterpenoid alkaloids reported in a JNP paper (2012, 75, 1145–1159). Tetrahedron 2014, 70, 2582–2590. [Google Scholar] [CrossRef]
- Güntzel, P.; Schilling, K.; Hanio, S.; Schlauersbach, J.; Schollmayer, C.; Meinel, L.; Holzgrabe, U. Bioinspired Ion Pairs Transforming Papaverine into a Protic Ionic Liquid and Salts. ACS Omega 2020, 5, 19202–19209. [Google Scholar] [CrossRef]
- Wu, L.X.; Xu, X.D.; Chen, X.; Miao, C.P.; Chen, Y.W.; Xu, L.H.; Zhao, L.X.; Li, Y.Q. Indole and tyramine alkaloids produced by an endophytic actinomycete associated with Artemisia annua. Chem. Nat. Compd. 2017, 53, 999–1001. [Google Scholar] [CrossRef]
- Wright, B.D.; Deblock, M.C.; Wagers, P.O.; Duah, E.; Robishaw, N.K.; Shelton, K.L.; Southerland, M.R.; DeBord, M.A.; Kersten, K.M.; McDonald, L.J.; et al. Anti-tumor activity of lipophilic imidazolium salts on select NSCLC cell lines. Med. Chem. Res. 2015, 24, 2838–2861. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | ||||
|---|---|---|---|---|---|---|---|
| δC, Type | δH, (J in Hz) | δH, (J in Hz) a | δC, Type | δH, (J in Hz) | δC, Type | δH, (J in Hz) | |
| 2 | 144.8, C | 144.7, C | 144.7, C | ||||
| 4 | 125.5, C | 125.5, C | 125.5, C | ||||
| 5 | 125.5, C | 125.5, C | 125.5, C | ||||
| 6 | 23.8, CH2 | 2.41, t (8.0) | 2.20, m | 23.8, CH2 | 2.40, t (7.8) | 23.7, CH2 | 2.30, t (8.1) |
| 7 | 20.6, CH2 | 1.34, m | 1.25, m | 20.6, CH2 | 1.34, m | 20.5, CH2 | 1.29, m |
| 8 | 13.3, CH3 | 0.80, t (7.3) | 0.69, t (7.3) | 13.3, CH3 | 0.80, t (7.2) | 13.2, CH3 | 0.71, t (7.3) |
| 9 | 7.9, CH3 | 2.15, s | 2.03, s | 7.9, CH3 | 2.13, s | 8.0, CH3 | 2.22, s |
| 10 | 8.0, CH3 | 2.20, s | 2.10, s | 8.0, CH3 | 2.19, s | 8.0, CH3 | 2.22, s |
| 1′ NH | 11.11, s | 10.80, s | 11.02, s | 11.03, s | |||
| 2′ | 123.9, CH | 7.17, s b | 7.02, s b | 123.8, CH | 7.16, s | 123.8, CH | 7.16, s |
| 3′ | 109.1, C | 109.1, C | 109.1, C | ||||
| 4′ | 126.8, C | 126.8, C | 126.8, C | ||||
| 5′ | 117.5, CH | 7.39, d (7.3) | 7.27, d (7.9) | 117.5, CH | 7.40, d (7.9) | 117.5, CH | 7.38, m |
| 6′ | 118.6, CH | 7.00, t (7.7) | 6.93, d (7.5) | 118.6, CH | 7.00, t (7.4) | 118.6, CH | 7.01, t (7.5) |
| 7′ | 121.2, CH | 7.09, t (7.5) | 7.02, m b | 121.2, CH | 7.09, t (7.4) | 121.2, CH | 7.09, t (7.5) |
| 8′ | 111.6, CH | 7.38, d (7.6) | 7.32, d (8.1) | 111.6, CH | 7.38, d (8.0) | 111.6, CH | 7.38, m |
| 9′ | 136.1, C | 136.1, C | 136.1, C | ||||
| 10′ | 25.0, CH2 | 3.08, t (7.0) | 3.03, t (6.8) | 25.0, CH2 | 3.07, t (6.7) | 24.9, CH | 2.95, t (7.2) |
| 11′ | 45.8, CH2 | 4.30, t (7.0) | 4.21, t (6.7) | 45.7, CH2 | 4.29, t (6.7) | 45.6, CH | 4.23, t (7.3) |
| 1″ c | 136.8, C | 126.7, C | 11.03, s | ||||
| 2″ | 128.9, CH | 7.16, m b | 7.02, m b | 129.9, CH | 6.91, d (7.9) | 123.8, CH | 7.16, s |
| 3″ | 128.6, CH | 7.32, t (7.3) | 7.21, m | 115.3, CH | 6.70, d (8.0) | 109.1, C | |
| 4″ | 127.0, CH | 7.28, m | 7.17, m | 156.5, C | 126.8, C | ||
| 5″ | 128.6, CH | 7.32, t (7.3) | 7.21, m | 115.3, CH | 6.70, d (8.0) | 117.5, CH | 7.38, m |
| 6″ | 128.9, CH | 7.16, m b | 7.02, m b | 129.9, CH | 6.91, d (7.9) | 118.6, CH | 7.01, t (7.5) |
| 7″ | 35.0, CH2 | 2.80, t (7.5) | 2.70, t (7.3) | 34.2, CH2 | 2.69, t (6.9) | 121.2, CH | 7.09, t (7.5) |
| 8″ | 45.8, CH2 | 4.22, t (7.5) | 4.09, t (7.2) | 46.2, CH2 | 4.14, t (6.9) | 111.6, CH | 7.38, m |
| 9″ | 136.1, C | ||||||
| 10″ | 24.9, CH | 2.95, t (7.2) | |||||
| 11″ | 45.6, CH | 4.23, t (7.3) | |||||
| Position | 4 | 5 | 6 | |||
|---|---|---|---|---|---|---|
| δC, Type | δH, (J in Hz) | δC, Type a | δH, (J in Hz) | δC, Type | δH, (J in Hz) | |
| 2 | 144.8, C | 144.8, C | 146.0, C | |||
| 4 | 125.5, C | 130.5, C | 125.5, C | |||
| 5 | 125.6, C | 125.7, C | 125.5, C | |||
| 6 | 23.9, CH2 | 2.53, t (7.9) | 23.8, CH2 | 2.55, m | 16.1, CH2 | 2.67, q (7.6) |
| 7 | 20.7, CH2 | 1.40, m | 20.4, CH2 | 1.43, m | 11.8, CH3 | 1.02, t (7.6) |
| 8 | 13.4, CH3 | 0.89, t (7.2) | 13.1, CH3 | 0.90, t (7.2) | 7.9, CH3 | 2.11, s |
| 9 | 7.9, CH3 | 2.10 or 2.11, s | 15.1, CH2 | 2.57, q (7.6) | 7.9, CH3 | 2.11, s |
| 10 | 7.9, CH3 | 2.10 or 2.11, s | 13.3, CH3 | 1.07, td (7.5, 1.9) | ||
| 11 | 7.6, CH3 | 2.10, d (1.5) | ||||
| 1′ | 136.9, C | 136.8, C | 136.9, C | |||
| 2′ | 129.0, CH | 7.19, d (7.2) | 128.7, CH | 7.20 or 7.17, d (7.1) | 129.0, CH | 7.19, d (7.1) |
| 3′ | 128.6, CH | 7.33, t (7.3) | 128.4, CH | 7.33, m | 128.6, CH | 7.33, t (7.3) |
| 4′ | 127.0, CH | 7.28, t (7.2) | 126.8, CH | 7.29, m | 127.0, CH | 7.28, t (7.3) |
| 5′ | 128.6, CH | 7.33, t (7.3) | 128.4, CH | 7.33, m | 128.6, CH | 7.33, t (7.3) |
| 6′ | 129.0, CH | 7.19, d (7.2) | 128.7, CH | 7.20 or 7.17, d (7.1) | 129.0, CH | 7.19, d (7.1) |
| 7′ | 35.0, CH2 | 2.93, t (7.0) | 35.2, CH2 | 2.93, m | 35.0, CH2 | 2.95, t (7.3) |
| 8′ | 46.0, CH2 | 4.28, t (7.0) | 45.6, CH2 | 4.29, m | 45.9, CH2 | 4.28, t (7.3) |
| 1″ | 126.5, C | 126.4, C | 136.9, C | |||
| 2″ | 129.9, CH | 6.92, d (8.1) | 129.6, CH | 6.93 or 6.90, d (8.2) | 129.0, CH | 7.19, d (7.1) |
| 3″ | 115.4, CH | 6.70, d (8.1) | 115.2, CH | 6.71 or 6.70, d (8.4) | 128.6, CH | 7.33, t (7.3) |
| 4″ | 156.8, C | 156.7, C | 127.0, CH | 7.28, t (7.3) | ||
| 5″ | 115.4, CH | 6.70, d (8.1) | 115.2, CH | 6.71 or 6.70, d (8.4) | 128.6, CH | 7.33, t (7.3) |
| 6″ | 129.9, CH | 6.92, d (8.1) | 129.6, CH | 6.93 or 6.90, d (8.2) | 129.0, CH | 7.19, d (7.1) |
| 7″ | 34.2, CH2 | 2.81, t (6.8) | 34.3, CH2 | 2.81, m | 35.0, CH2 | 2.95, t (7.3) |
| 8″ | 46.4, CH2 | 4.21, t (6.9) | 46.0, CH2 | 4.21, m | 45.9, CH2 | 4.28, t (7.3) |
| Position | δC, Type | δH, (J in Hz) | Position | δC, Type | δH, (J in Hz) |
|---|---|---|---|---|---|
| 2 | 153.1, C | 1′ NH | 11.06, s | ||
| 3 | 142.6, C | 2′ | 124.2, CH | 7.13, s | |
| 4 | 144.5, CH | 8.20, s | 3′ | 108.3, C | |
| 5 | 140.6, C | 4′ | 126.8, C | ||
| 6 | 142.6, CH | 8.40, s | 5′ | 117.3, CH | 7.25, d (7.9) |
| 7 | 29.5, CH2 | 2.83, t (8.2) | 6′ | 118.6, CH | 6.91, t (7.4) |
| 8 | 21.8, CH2 | 1.56, m | 7′ | 121.2, CH | 7.06, t (7.3) |
| 9 | 13.8, CH3 | 1.00, t (7.0) | 8′ | 111.6, CH | 7.35, d (8.1) |
| 10 | 24.5, CH2 | 2.73, q (7.5) | 9′ | 136.0, C | |
| 11 | 14.4, CH3 | 1.13, t (7.5) | 10′ | 26.5, CH2 | 3.35, m a |
| 12 | 24.5, CH2 | 2.57, q (7.5) | 11′ | 58.5, CH2 | 4.78, t (6.5) |
| 13 | 13.9, CH3 | 1.01, t (7.5) |
| Test organism | MIC (μg/mL, n = 3) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | Rifampicin | Tetracycline | Gentamicin | |
| B. subtilis DSM10 | 8 | >32 | 8 | >32 | 32 | 32 | >32 | <0.031 | 2–4 | 0.06 |
| M. smegmatis ATCC607 | 4 | >32 | 4 | >32 | >32 | 16 | >32 | 8–16 | 0.25–0.5 | 4–8 a |
| L. monocytogenes DSM20600 | 32 | >32 | 8 | >32 | >32 | >32 | >32 | <0.031 | 0.5–1 | <0.031 |
| S. aureus ATCC25923 | 16 | >32 | 8 | >32 | >32 | 32 | >32 | <0.031 | 0.25–0.5 | 0.06–0.125 |
| E. coli ATCC25922 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | 4 | 2–4 | 0.06 |
| Nystatin | Tebuconazole | Amphotericin B | ||||||||
| C. albicans FH2173 b | >32 | >32 | 16 | >32 | >32 | >32 | >32 | 1–2 | 0.25 | 0.5–1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Marner, M.; Mettal, U.; Liu, Y.; Schäberle, T.F. Seven New Alkaloids Isolated from Marine Flavobacterium Tenacibaculum discolor sv11. Mar. Drugs 2022, 20, 620. https://doi.org/10.3390/md20100620
Wang L, Marner M, Mettal U, Liu Y, Schäberle TF. Seven New Alkaloids Isolated from Marine Flavobacterium Tenacibaculum discolor sv11. Marine Drugs. 2022; 20(10):620. https://doi.org/10.3390/md20100620
Chicago/Turabian StyleWang, Lei, Michael Marner, Ute Mettal, Yang Liu, and Till F. Schäberle. 2022. "Seven New Alkaloids Isolated from Marine Flavobacterium Tenacibaculum discolor sv11" Marine Drugs 20, no. 10: 620. https://doi.org/10.3390/md20100620