
Cellular Signal Transductions and Their Inhibitors Derived from Deep-Sea Organisms


Abstract
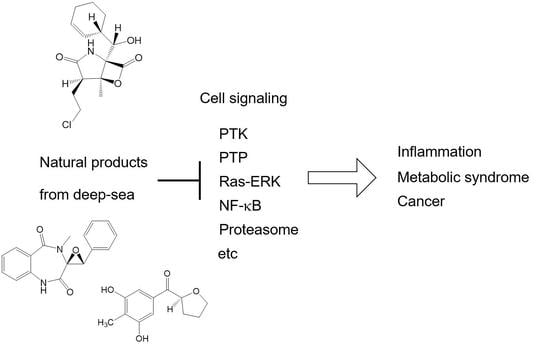
:
1. Introduction
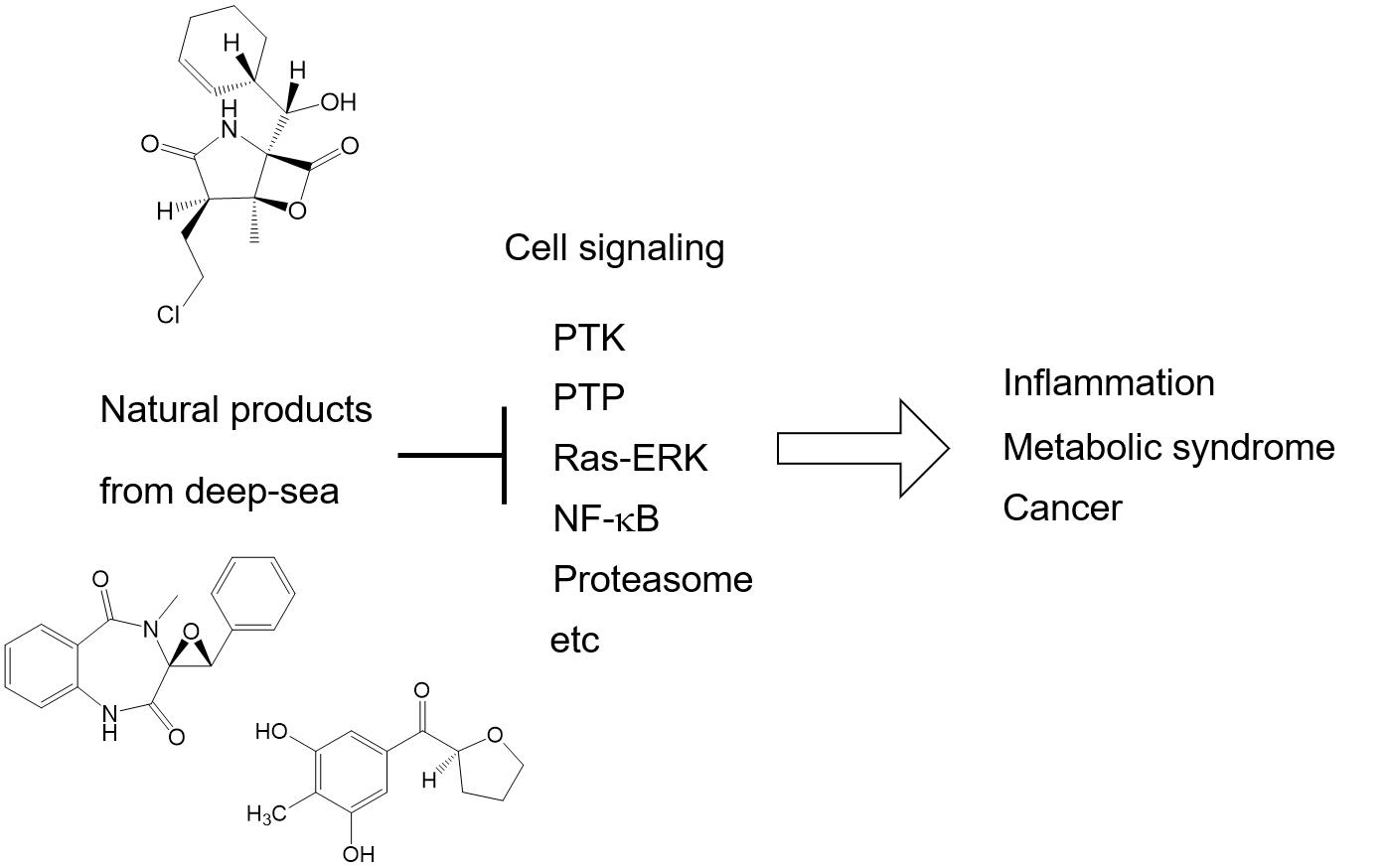
2. Cellular Signal Transduction and Alteration in Disease
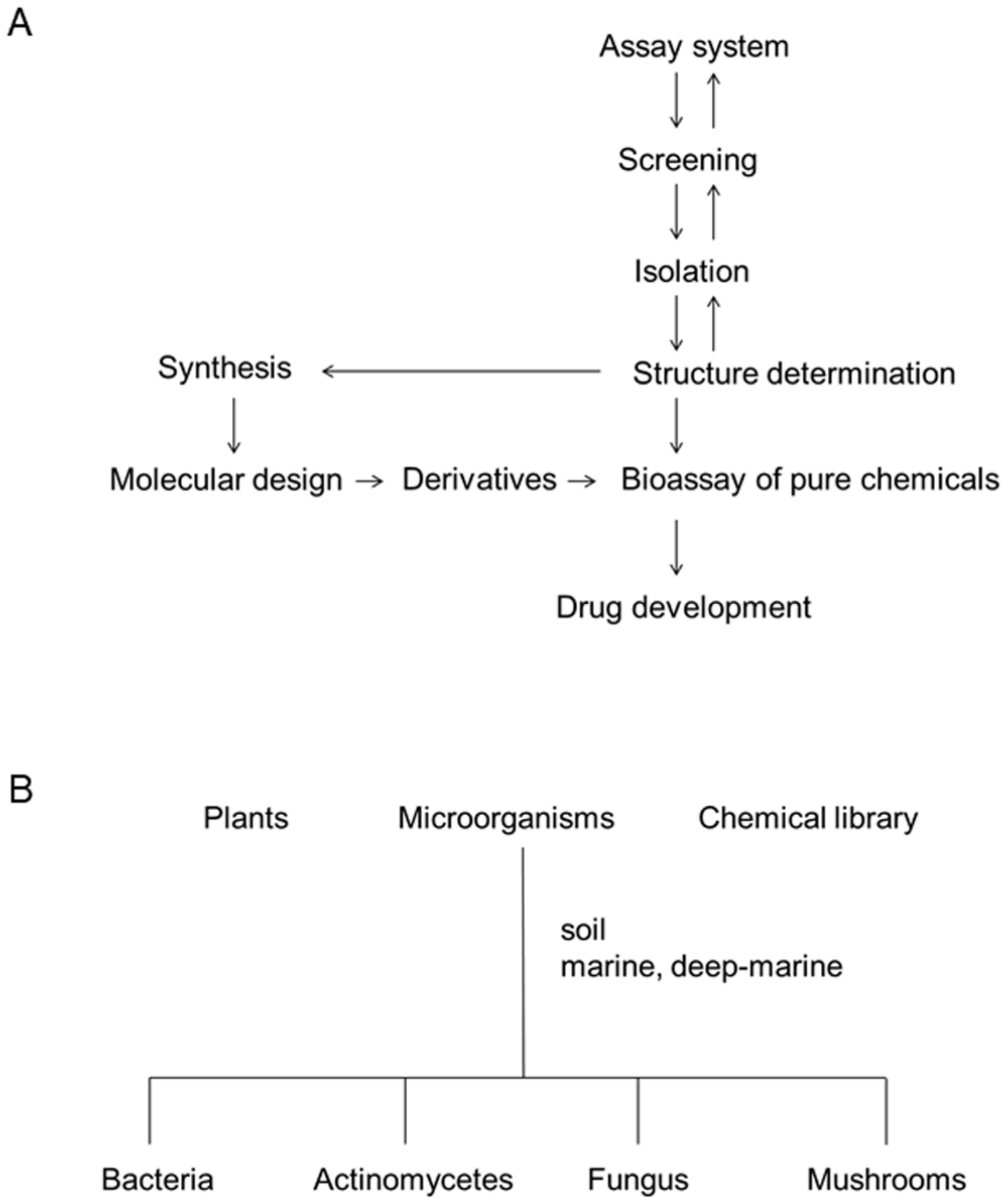
3. Process of Screening and Deep-Sea Organisms as a Source of Bioactive Metabolites
4. Anti-Inflammatory Agents from Deep-Sea Organisms
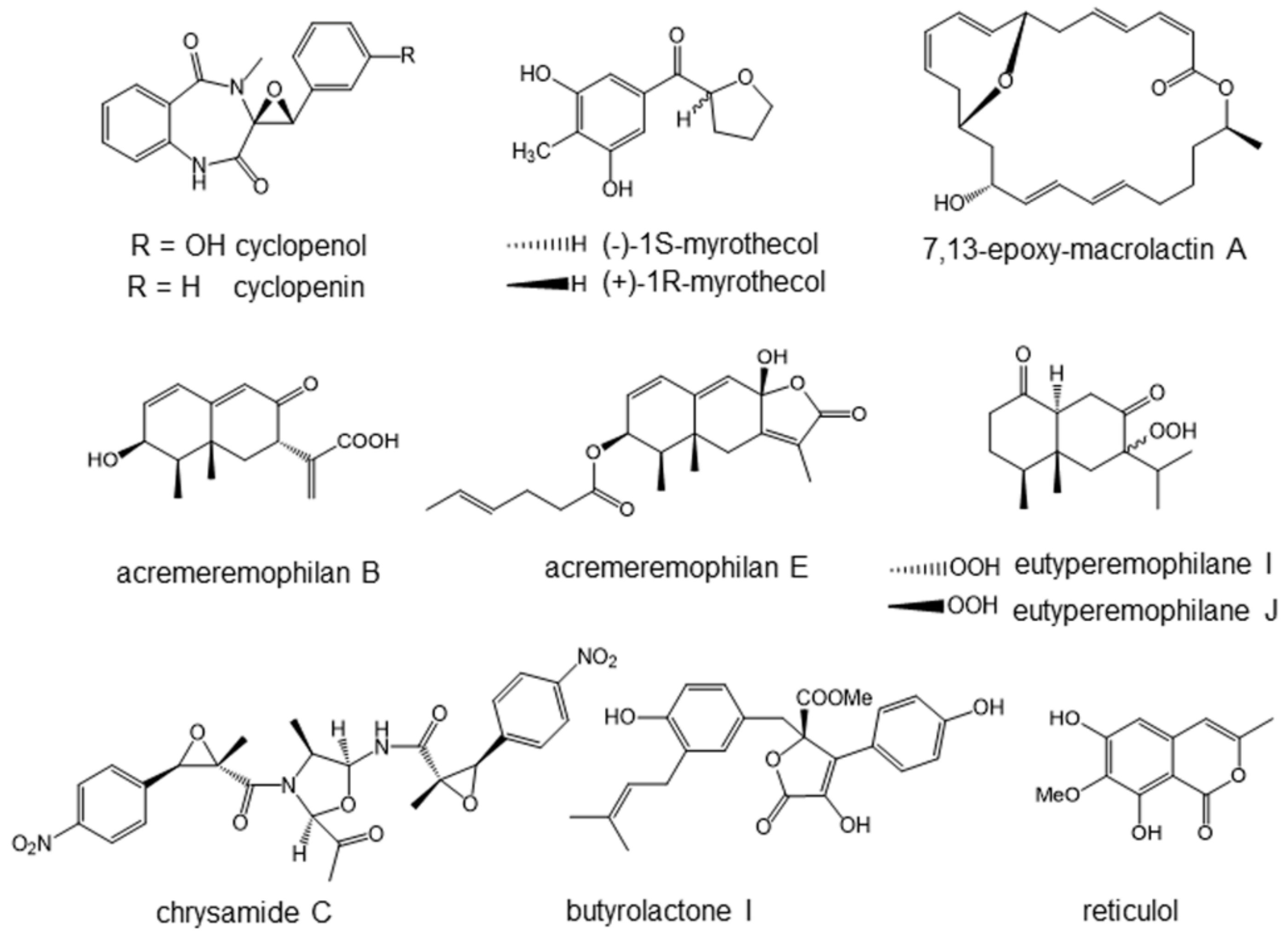
4.1. Cyclopenol and Cyclopenin Inhibiting NF-κB Signaling
4.2. Myrothenols Inhibiting LPS-Induced NO Production
4.3. Macrolactins Inhibiting NO and Cytokine Productions
4.4. Acremeremophilanes Inhibiting LPS-Induced NO Production
4.5. Eutyperemophilanes Inhibiting LPS-Induced NO Production
4.6. Chrysamide C Inhibiting Interleukin-17 Production
4.7. Butyrolactone I Suppressing Mast Cell Activity
4.8. Reticurol Suppressing Mast Cell Activity
5. Modulators of Metabolic Syndrome Model and Antimicrobial Compounds
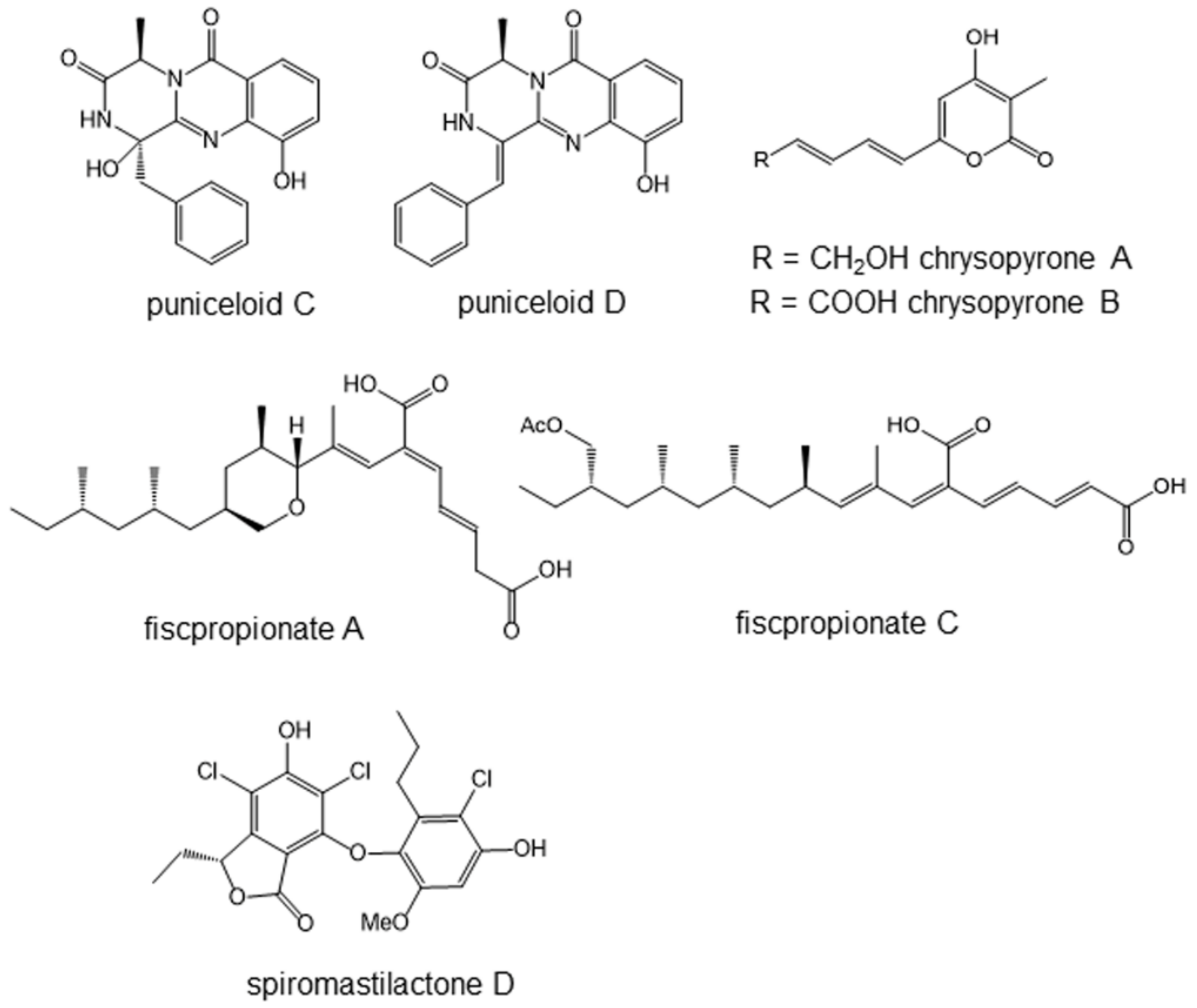
5.1. Puniceloids C and D, Liver X Receptor Agonists
5.2. Chrysopyrones A and B, Protein–Tyrosine Phosphatase Inhibitors That Ameliorate Diabetes Mellitus Model
5.3. Fiscpropionate A and C Inhibiting Bacterial Protein–Tyrosine Phosphatase
5.4. Spiromastilactone D Inhibits Influenza Virus Replication
6. Anticancer Agents
6.1. Cytotoxic Agents and Cell Signaling Inhibitors
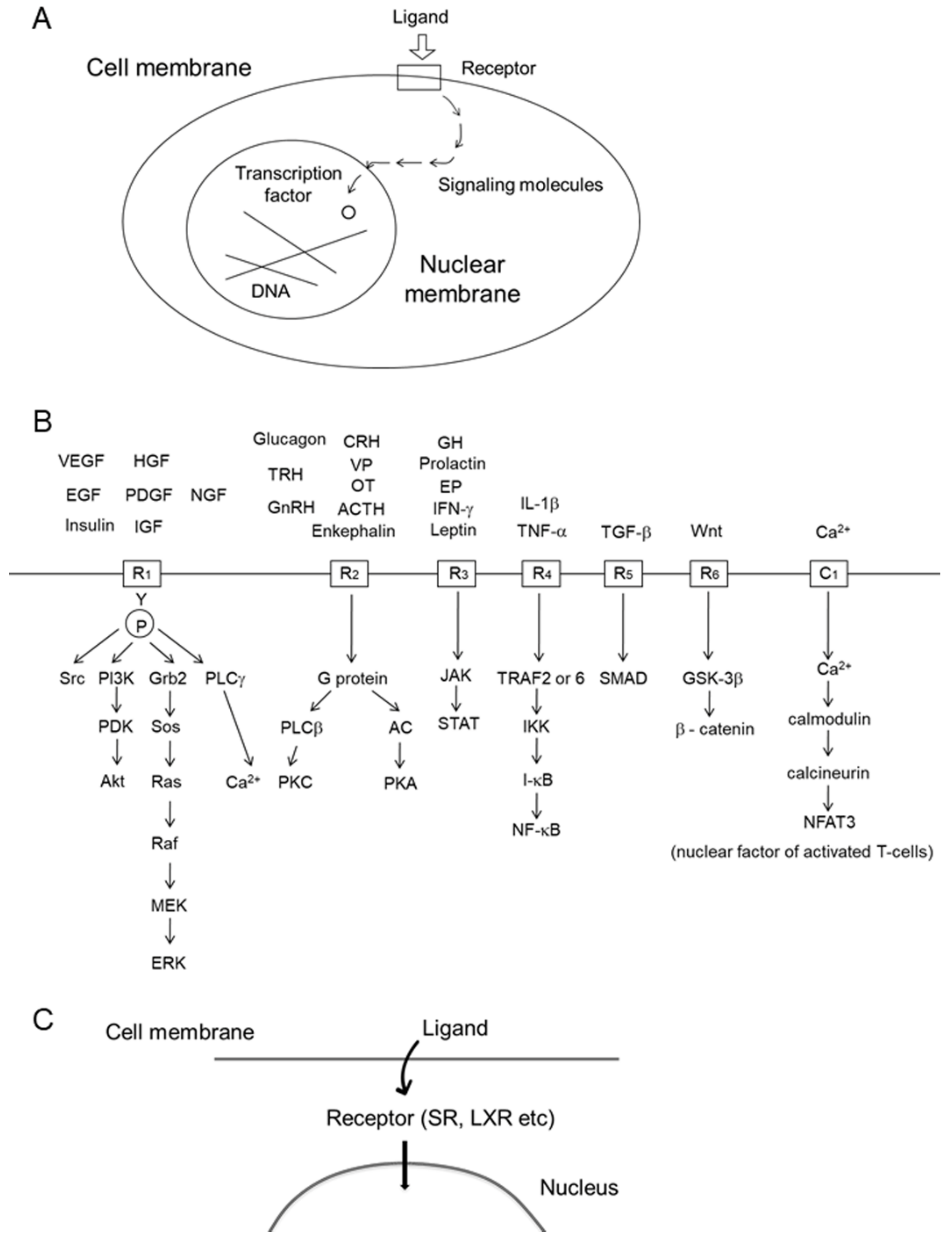
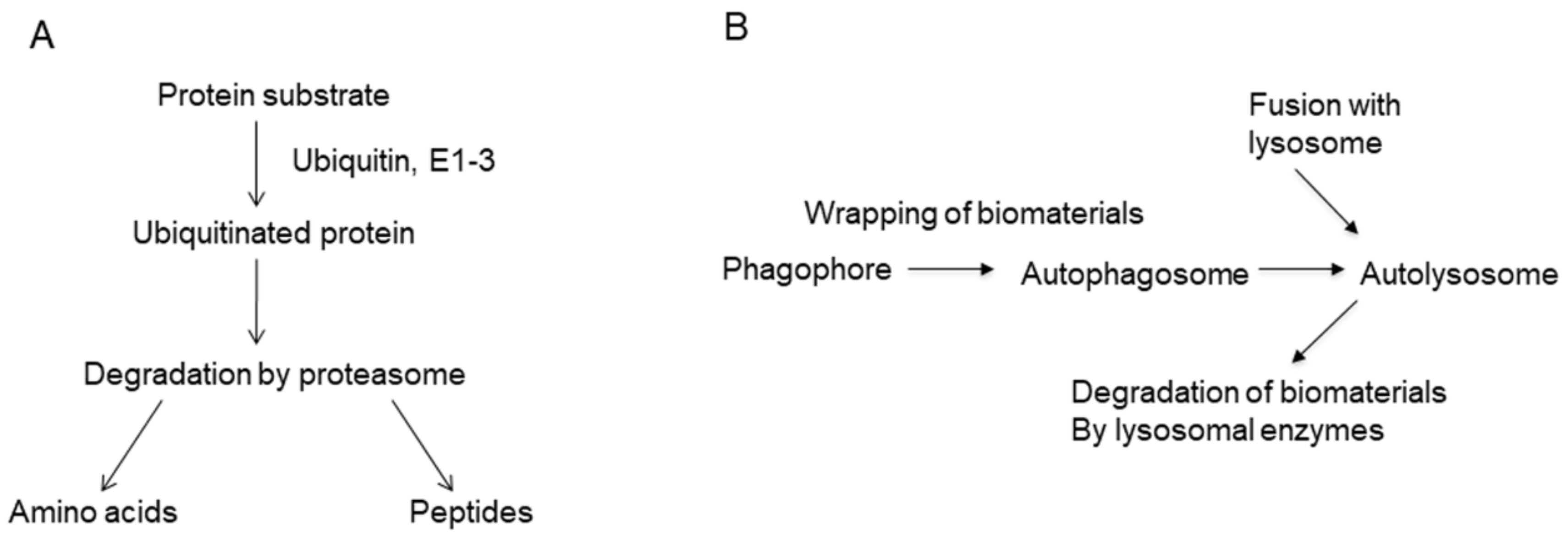
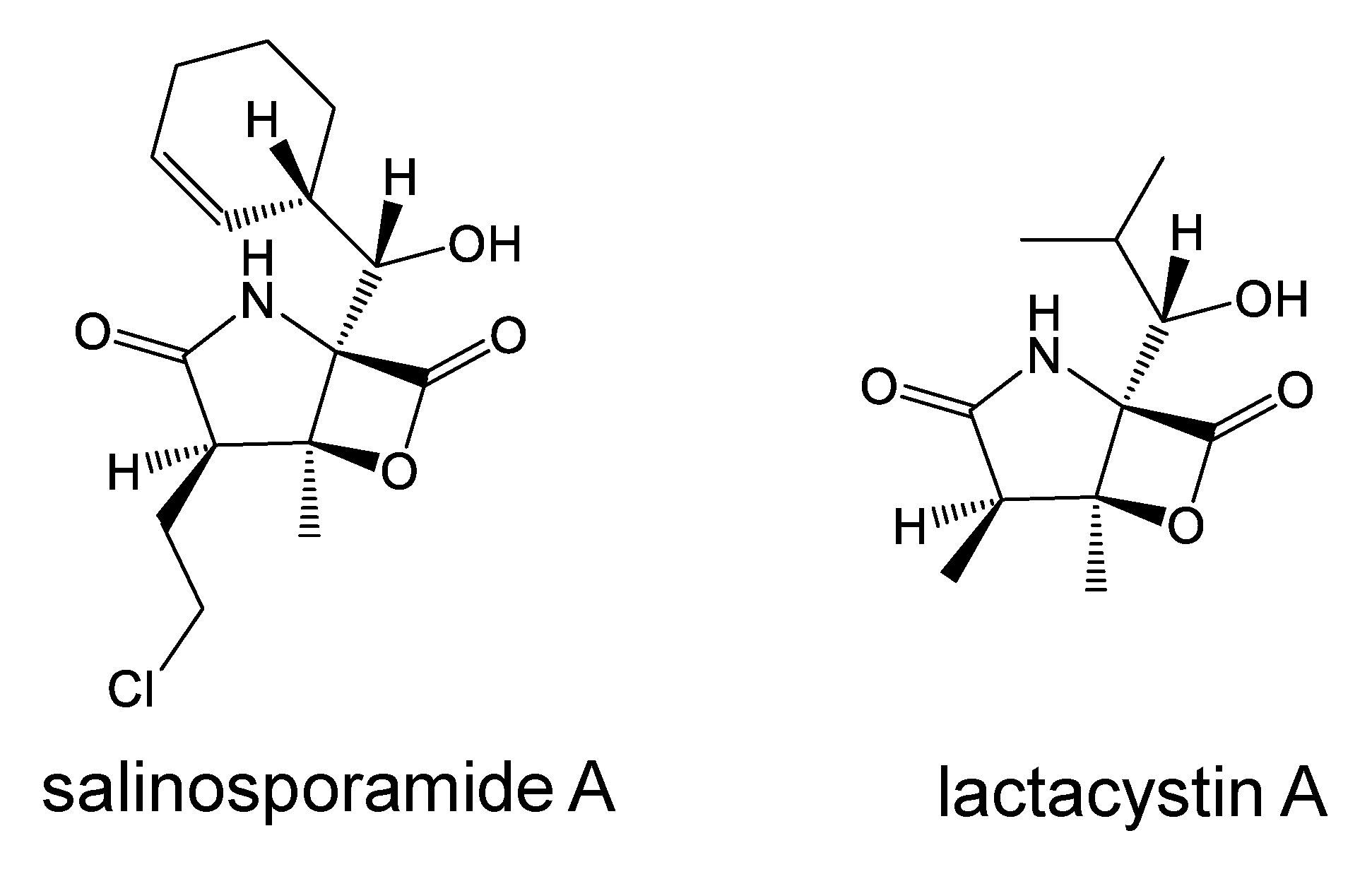
6.2. Salinosporamide A, a Proteasome Inhibitor
7. Conclusions and Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rall, T.W.; Sutherland, E.W. Formation of a cyclic adenine ribonucleotide by tissue particles. J. Biol. Chem. 1958, 232, 1065–1076. [Google Scholar] [CrossRef]
- Onoda, T.; Iinuma, H.; Sasaki, Y.; Hamada, M.; Isshiki, K.; Naganawa, H.; Takeuchi, T.; Tatsuta, K.; Umezawa, K. Isolation of a novel tyrosine kinase inhibitor, lavendustin A, from Streptomyces griseolavendus. J. Nat. Prod. 1989, 52, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
- Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discov. 2002, 1, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Imoto, M.; Kakeya, H.; Sawa, T.; Hayashi, C.; Hamada, M.; Takeuchi, T.; Umezawa, K. Dephostasin, a novel protein tyrosine phosphatase inhibitor produced by Streptomyce I. Taxonomy, isolation, and characterization. J. Antibiot. 1993, 46, 1342–1346. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Hiroki, A.; Watanabe, T.; Yamashita, T.; Takei, I.; Umezawa, K. Potentiation of insulin-related signal transduction by a novel protein-tyrosine phosphatase inhibitor, Et-3,4-dephostatin, on cultured 3T3-L1 adipocytes. J. Biol. Chem. 2001, 276, 27511–27518. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, N.; Ariga, A.; To-e, S.; Nakamura, H.; Agata, N.; Hirano, S.; Inoue, J.; Umezawa, K. Synthesis of NF-κB activation inhibitors derived from epoxyquinomicin C. Bioorg. Med. Chem. Lett. 2000, 10, 865–869. [Google Scholar] [CrossRef]
- Ariga, A.; Namekawa, J.; Matsumoto, N.; Inoue, J.; Umezawa, K. Inhibition of TNF-κ-induced nuclear translocation and activation of NF-B by dehydroxymethyl-epoxyquinomicin. J. Biol. Chem. 2002, 277, 27625–27630. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Gao, X.; Wang, X.; Li, X.; Jiang, X.; Xie, Z.; Ma, K.; Ma, J.; Umezawa, K.; Zhang, Y. Comparison of anti-atopic dermatitis activities between DHMEQ and tacrolimus ointments in mouse model without stratum corneum. Int. Immunopharm. 2019, 71, 43–51. [Google Scholar] [CrossRef]
- Umezawa, K.; Breborowicz, A.; Gantsev, G. Anticancer activity of novel NF-B inhibitor DHMEQ by intraperitoneal administration. Oncol. Res. 2020, 28, 541–550. [Google Scholar] [CrossRef]
- Liu, S.; Ren, J.; Dijke, P.T. Targeting TGFβ signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 8. [Google Scholar] [CrossRef]
- Atsumi, S.; Nagasawa, A.; Koyano, T.; Kowithayakorn, T.; Umezawa, K. Suppression of TGF-β signaling by conophylline via upregulation of c-Jun expression. Cell. Mol. Life Sci. 2003, 60, 2516–2525. [Google Scholar] [CrossRef]
- Liu, J.; Farmer, J.D., Jr.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 65, 807–815. [Google Scholar] [CrossRef]
- Welchman, R.L.; Gordon, C.; Mayer, R.J. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell Biol. 2005, 6, 599–609. [Google Scholar] [CrossRef]
- Paramore, A.; Franz, S. Bortezomib. Nat. Rev. Drug Discov. 2003, 2, 611–612. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Jing, K.; Lim, K. Why is autophagy important in human diseases? Exp. Mol. Med. 2012, 44, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Sasazawa, Y.; Sato, N.; Umezawa, K.; Simizu, S. Conophylline protects cells in cellular models of neurodegenerative diseases by inducing mTOR-independent autophagy. J. Biol. Chem. 2015, 290, 6168–6178. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, T.; Nakade, Y.; Ibusuki, M.; Kitano, R.; Yamauchi, T.; Kimoto, S.; Inoue, T.; Kobayashi, Y.; Ishii, N.; Sumida, Y.; et al. Conophylline inhibits high fat diet-induced non-alcoholic fatty liver disease in mice. PLoS ONE 2019, 14, e0210068. [Google Scholar] [CrossRef]
- Skropeta, D.; Wei, L. Recent advances in deep-sea natural products. Nat. Prod. Rep. 2014, 31, 999–1025. [Google Scholar] [CrossRef]
- Dumdei, E.J.; Blunt, J.W.; Munro, M.H.G.; Pannell, L.K. Isolation of Calyculins, Calyculinamides, and Swinholide H from the New Zealand Deep-Water Marine Sponge Lamellomorpha strongylata. J. Org. Chem. 1997, 62, 2636–2639. [Google Scholar] [CrossRef]
- Schupp, P.J.; Kohlert-Schupp, C.; Whitefield, S.; Engemann, A.; Rohde, S.; Hemscheidt, T.; Pezzuto, J.M.; Kondratyuk, T.P.; Park, E.J.; Marler, L.; et al. Cancer chemopreventive and anticancer evaluation of extracts and fractions from marine macro- and micro-organisms collected from Twilight Zone waters around Guam. Nat. Prod. Commun. 2009, 4, 1717–1728. [Google Scholar]
- Wright, A.D.; Schupp, P.J.; Schror, J.P.; Engemann, A.; Rohde, S.; Kelman, D.; Voogd-de, N.; Carroll, A.; Motti, C.A. Twilight zone sponges from Guam yield theonellin isocyanate and psammaplysins I and J. J. Nat. Prod. 2012, 75, 502–506. [Google Scholar] [CrossRef]
- Wang, L.; Li, M.; Lin, Y.; Du, S.; Liu, Z.; Ju, J.; Suzuki, H.; Sawada, M.; Umezawa, K. Inhibition of cellular inflammatory mediator production and amelioration of learning deficit in flies by deep sea Aspergillus-derived cyclopenin. J. Antibiot. 2020, 73, 622–629. [Google Scholar] [CrossRef]
- Bracken, A.; Pocker, A.; Raistrick, H. Studies in the biochemistry of microorganisms. Cyclopenin, a nitrogen-containing metabolic product of Penicillium cyclopium Westling. Biochem. J. 1954, 57, 587–595. [Google Scholar] [CrossRef] [Green Version]
- Birkinshaw, J.H.; Luckner, M.; Mohammed, Y.S.; Mothes, K.; Stickings, C.E. Studies in the biochemistry of microorganisms. 114. Viridicatol and cyclopenol, metabolites of Penicullium viridicatum Westling and Penicillium cyclopium Westling. Biochem. J. 1963, 89, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; He, J.; Wu, Y.; Du, N.; Li, X.; Ju, J.; Hu, Z.; Umezawa, K.; Wang, L. Isolation and characterization of New anti-inflammatory and antioxidant components from deep marine-derived fungus Myrothecium sp. Bzo-1062. Mar. Drugs 2020, 18, 597. [Google Scholar] [CrossRef]
- Yan, X.; Zhou, Y.X.; Tang, X.X.; Liu, X.X.; Yi, Z.W.; Fang, M.J.; Wu, Z.; Jiang, F.Q.; Qiu, Y.K. Macrolactins from marine-derived Bacillus subtilis B5 bacteria as inhibitors of inducible nitric oxide and cytokines expression. Mar. Drugs 2016, 14, 195. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Zhao, J.; Liu, D.; Proksch, P.; Zhao, Z.; Lin, W. Eremophilane-type Sesquiterpenoids from an Acremonium sp. fungus isolated from deep-sea sediments. J. Nat. Prod. 2016, 79, 1035–1047. [Google Scholar] [CrossRef]
- Niu, S.; Liu, D.; Shao, Z.; Proksch, P.; Lin, W. Eremophilane-type sesquiterpenoids in a deep-sea fungus Eutypella sp. activated by chemical epigenetic manipulation. Tetrahedron 2018, 74, 7310–7325. [Google Scholar] [CrossRef]
- Chen, S.; Wang, J.; Lin, X.; Zhao, B.; Wei, X.; Li, G.; Kaliaperumal, K.; Liao, S.; Yang, B.; Zhou, X.; et al. Chrysamides A–C, three dimeric nitrophenyl trans-epoxyamides produced by the deep-sea-derived fungus Penicillium chrysogenum SCSIO41001. Org. Lett. 2016, 18, 3650–3653. [Google Scholar] [CrossRef]
- Metcalfe, D.D.; Baram, D.; Mekori, Y.A. Mast cells. Physiol. Rev. 1997, 77, 1033–1079. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.F.; Lockey, R.F. Anaphylaxis: A review of causes and mechanisms. J. Allergy Clin. Immunol. 2002, 110, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Noma, N.; Asagiri, M.; Takeiri, M.; Ohmae, S.; Takemoto, K.; Iwaisako, K.; Simizu, S.; Umezawa, K. Inhibition of MMP-2-mediated mast cell invasion by NF-B inhibitor DHMEQ in mast cells. Int. Arch. Allergy Immunol. 2015, 166, 84–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.M.; Xie, C.L.; Gao, Y.Y.; Liu, B.; Lin, W.X.; Liu, H.; Cao, M.J.; Su, W.J.; Yang, X.W.; Liu, G.M. Deep-sea-derived butyrolactone I suppresses ovalbumin-induced anaphylaxis by regulating mast cell function in a murine model. J. Agric. Food Chem. 2018, 66, 5581–5592. [Google Scholar] [CrossRef]
- Niu, S.; Liu, Q.; Xia, J.M.; Xie, C.L.; Luo, Z.H.; Shao, Z.; Liu, G.; Yang, X.W. Polyketides from the deep-sea-derived fungus Graphostroma sp. MCCC 3A00421 showed potent antifood allergic activities. J. Agric. Food Chem. 2018, 66, 1369–1376. [Google Scholar] [CrossRef]
- Furutani, Y.; Shimada, M.; Hamada, M.; Takeuchi, T.; Umezawa, H. Reticulol, an inhibitor of cyclic nucleotide phosphodiesterases. J. Antibiot. 1975, 28, 558–560. [Google Scholar] [CrossRef] [Green Version]
- Lim, D.-S.; Kwak, Y.-S.; Lee, K.-H.; Ko, S.-H.; Yoon, W.-H.; Lee, W.; Kim, C. Topoisomerase I inactivation by reticulol and its in vivo cytotoxicity against B16F10 melanoma. Chemotherapy 2003, 49, 257–263. [Google Scholar] [CrossRef]
- Lim, D.-S.; Kwak, Y.-S.; Kim, J.-H.; Ko, S.-H.; Yoon, W.-H.; Kim, C.-H. Antitumor efficacy of reticulol from Streptoverticillium against the lung metastasis model B16F10 melanoma. Lung metastasis inhibition by growth inhibition of melanoma. Chemotherapy 2003, 49, 146–153. [Google Scholar] [CrossRef]
- Hong, C.; Tontonoz, P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. Rev. Drug Discov. 2014, 13, 433–444. [Google Scholar] [CrossRef]
- El-Gendy, B.E.-D.M.; Goher, S.S.; Hegazy, L.S.; Arief, M.M.H.; Burris, T.P. Recent Advances in the Medicinal Chemistry of Liver X Receptors: Miniperspective. J. Med. Chem. 2018, 61, 10935–10956. [Google Scholar] [CrossRef]
- Liang, X.; Zhang, X.; Lu, X.; Zheng, Z.; Ma, X.; Qi, S. Diketopiperazine-type alkaloids from a deep-sea-derived Aspergillus puniceus fungus and their effects on liver X receptor α. J. Nat. Prod. 2019, 82, 1558–1564. [Google Scholar] [CrossRef]
- Gum, R.J.; Gaede, L.L.; Koterski, S.L.; Heindel, M.; Clampit, J.E.; Zinker, B.A.; Trevillyan, J.M.; Ulrich, R.G.; Jirousek, M.R.; Rondinone, C.M. Reduction of protein tyrosine phosphatase 1B increases insulin-dependent signaling in ob/ob mice. Diabetes 2003, 52, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Han, T.W.; Cai, J.; Zhong, W.; Xu, G.; Wang, F.; Tian, X.; Zhou, X.; Liu, Q.; Liu, Y.; Wang, J. Protein tyrosine phosphatase 1B (PTP1B) inhibitors from the deep-sea fungus Penicillium chrysogenum SCSIO 07007. Bioorg. Chem. 2020. [Google Scholar] [CrossRef]
- Koul, A.; Herget, T.; Klebl, B.; Ullrich, A. Interplay between mycobacteria and host signaling pathways. Nat. Rev. Microbiol. 2004, 2, 189–202. [Google Scholar] [CrossRef]
- Zhou, B.; He, Y.; Zhang, X.; Xu, J.; Luo, Y.; Wang, Y.; Franzblau, S.G.; Yang, Z.; Chan, R.J.; Liu, Y.; et al. Targeting mycobacterium protein tyrosine phosphatase B for antituberculosis agents. Proc. Natl. Acad. Sci. USA 2010, 107, 4573–4578. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wang, Q.; Li, S.; Cui, H.; Sun, Z.; Chen, D.; Lu, Y.; Liu, H.; Zhang, W. Polypropionate derivatives with mycobacterium tuberculosis protein tyrosine phosphatase B inhibitory activities from the deep-sea-derived fungus Aspergillus fischeri FS452. J. Nat. Prod. 2019, 82, 3440–3449. [Google Scholar] [CrossRef]
- Niu, S.; Si, L.; Liu, D.; Zhou, A.; Zhang, Z.; Shao, Z.; Wang, S.; Zhang, L.; Zhou, D.; Lin, W. Spiromastilactones: A new class of influenza virus inhibitors from deep-sea fungus. Eur. J. Med. Chem. 2016, 108, 229–244. [Google Scholar] [CrossRef]
- Das, S.; Guha, I.; Chatterjee, A.; Banerji, A. Anti-cancer potential of all-trans retinoic acid (ATRA): A Review. Proc. Zool. Soc. 2013, 66, 1–7. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Lu, Z.; Hunter, T. Ubiquitylation and proteasomal degradation of the p21(Cip1), 27(Kip1) and p57(Kip2) CDK inhibitors. Cell Cycle 2010, 9, 2342–2352. [Google Scholar] [CrossRef] [Green Version]
- Love, I.M.; Shi, D.; Grossman, S.R. p53 ubiquitination and proteasomal degradation. Methods Mol. Biol. 2013, 962, 63–73. [Google Scholar] [PubMed]
- Feling, R.H.; Buchanan, G.O.; Mincer, T.J.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Salinosporamide A: A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angew. Chem. Int. Ed. 2003, 42, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Omura, S.; Fujimoto, T.; Otoguro, K.; Matsuzaki, K.; Moriguchi, R.; Tanaka, H.; Sasaki, Y. Lactacystin, a novel microbial metabolite, induces neuritogenesis of neuroblastoma cell. J. Antibiot. 1991, 44, 113–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groll, M.; Huber, R.; Potts, B.C.M. Crystal structures of salinosporamide A (NPI-0052) and B (NPI-0047) in complex with the 20S proteasome reveal important consequences of β-lactone ring opening and a mechanism for irreversible binding. J. Am. Chem. Soc. 2006, 128, 5136–5141. [Google Scholar] [CrossRef]
- Ahn, K.S.; Sethi, G.; Chao, T.-H.; Neuteboom, S.T.C.; Chaturvedi, M.M.; Palladino, M.A.; Younes, A.; Aggarwal, B.B. Salinosporamide A (NPI-0052) potentiates apoptosis, suppresses osteoclastogenesis, and inhibits invasion through down-modulation of NF-κB–regulated gene products. Blood 2007, 110, 2286–2295. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Natural Products | Target Signal | Related Illustration | Reference |
|---|---|---|---|
| Cyclopenol and cyclopenin | NO production (NF-κB) | Figure 1B, R4 | [23] |
| Myrothecols | NO production | Figure 1B, R4 | [26] |
| 7,13-Epoxyl-macrolactin A | NO production | Figure 1B, R4 | [27] |
| Acremeremophilane B | NO production | Figure 1B, R4 | [28] |
| Eutyperemophilane I and J | NO production | Figure 1B, R4 | [29] |
| Chrysamide C | IL-17 Production | __ * | [30] |
| Butyrolactone I | Mast cell activity | __ | [34] |
| Reticurol | Mast cell activity | __ | [35] |
| Puniceloids C and D | Liver X receptor | Figure 1C | [41] |
| Chrysopyrones A and B | Protein–tyrosine phosphatase | Figure 1B, R1 | [43] |
| Fiscpropionate A and C | Protein–tyrosine phosphatase (bacterial) | __ | [46] |
| Spiromastilactone D | Influenza virus | __ | [47] |
| Salinosporamide A | Proteasome | Figure 2A | [52] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Umezawa, K. Cellular Signal Transductions and Their Inhibitors Derived from Deep-Sea Organisms. Mar. Drugs 2021, 19, 205. https://doi.org/10.3390/md19040205
Wang L, Umezawa K. Cellular Signal Transductions and Their Inhibitors Derived from Deep-Sea Organisms. Marine Drugs. 2021; 19(4):205. https://doi.org/10.3390/md19040205
Chicago/Turabian StyleWang, Liyan, and Kazuo Umezawa. 2021. "Cellular Signal Transductions and Their Inhibitors Derived from Deep-Sea Organisms" Marine Drugs 19, no. 4: 205. https://doi.org/10.3390/md19040205