
Structure Revision and Protein Tyrosine Phosphatase Inhibitory Activity of Drazepinone


Abstract
:
1. Introduction
2. Results and Discussion
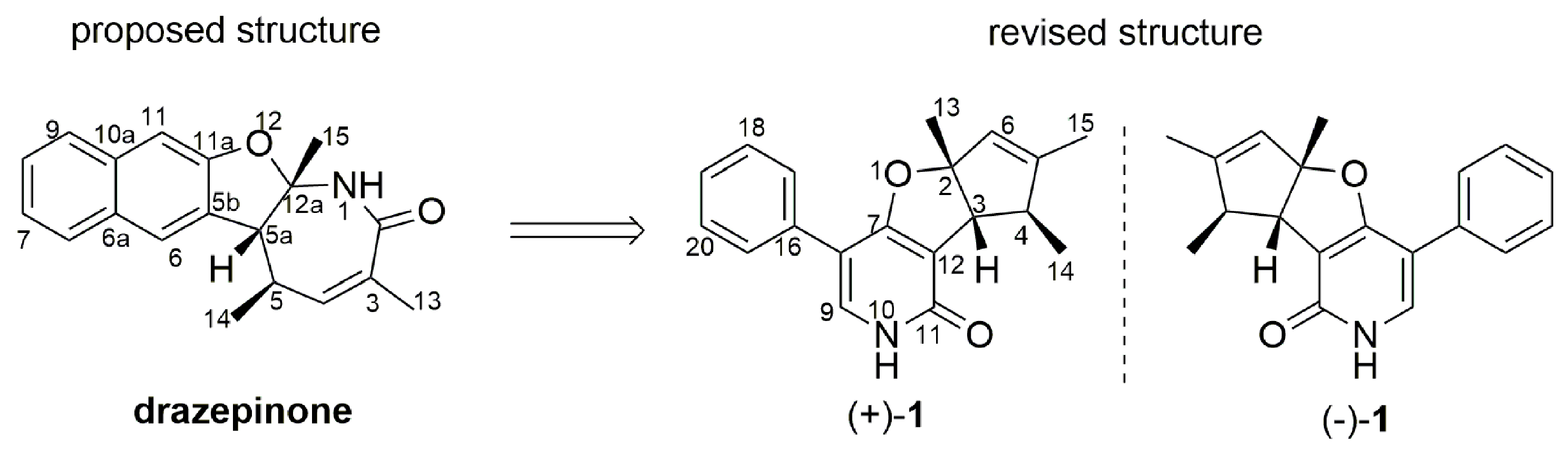
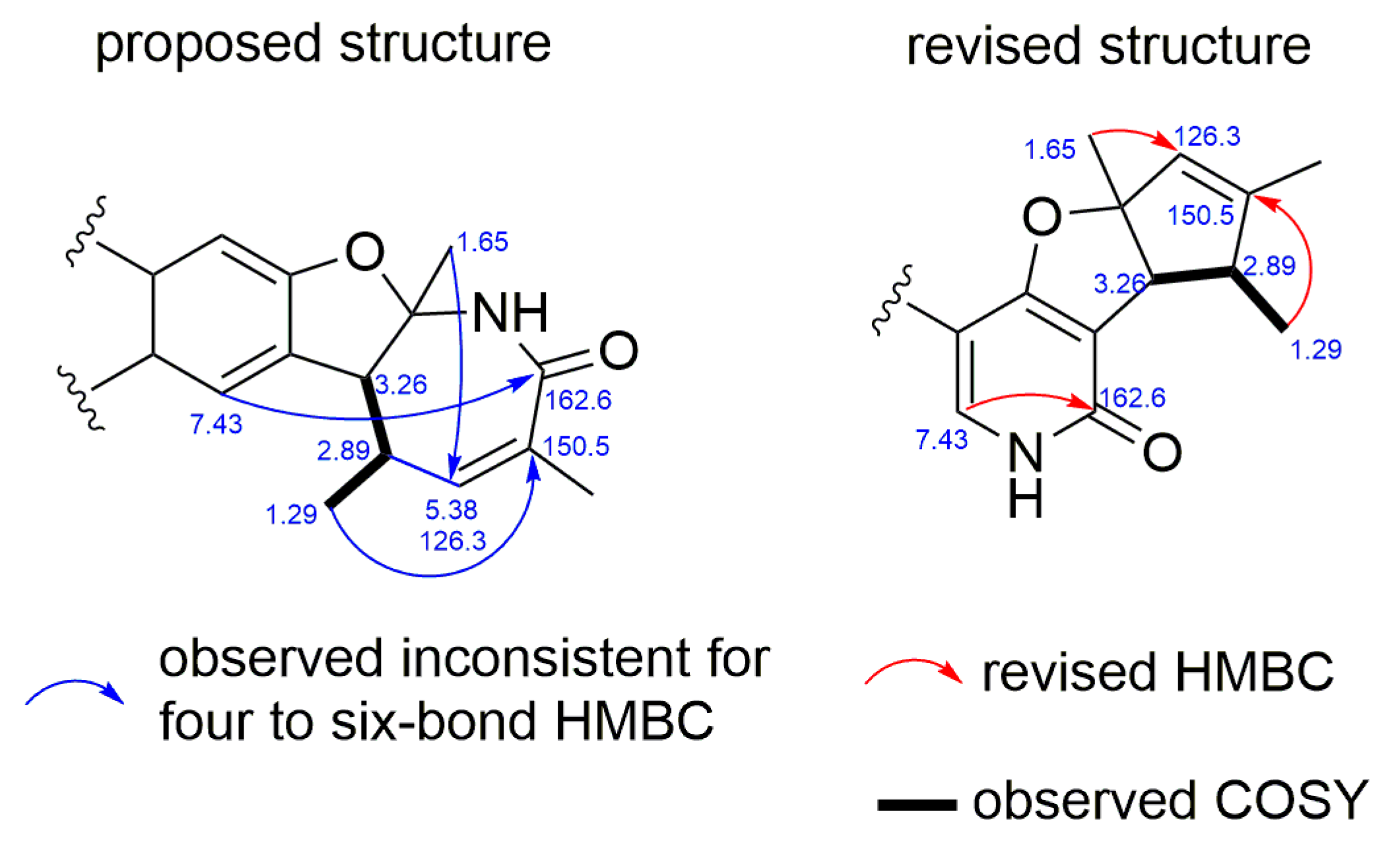
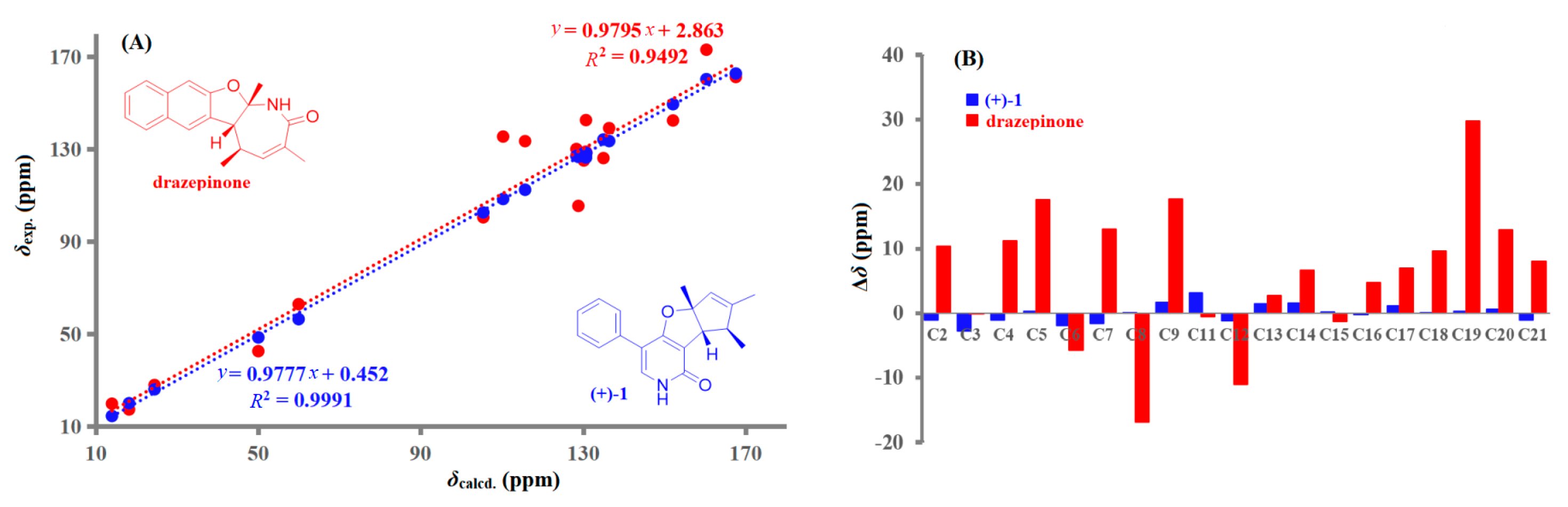
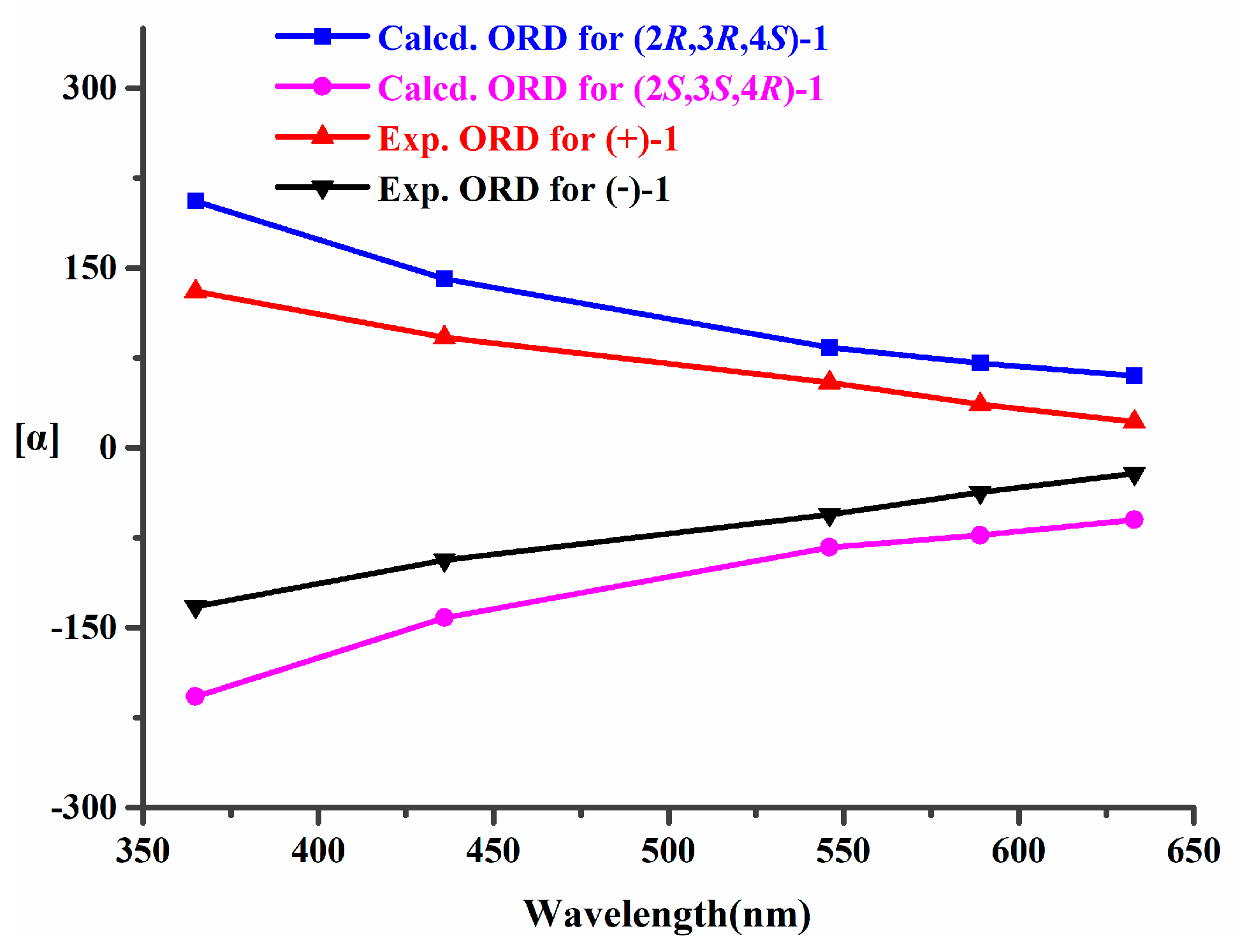
2.1. Structural Elucidation and Revision
2.2. PTP Inhibitory Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Isolation of Fungal Material
3.3. Computational Section
3.4. Enzyme Inhibitory Activity Assay
3.5. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nicolaou, K.C.; Snyder, S.A. Chasing molecules that were never there: Misassigned natural products and the role of chemical synthesis in modern structure elucidation. Angew. Chem. Int. Ed. 2005, 44, 1012–1044. [Google Scholar] [CrossRef]
- Evidente, A.; Andolfi, A.; Vurro, M.; Fracchiolla, M.; Zonno, M.C.; Motta, A. Drazepinone, a trisubstituted tetrahydronaphthofuroazepinone with herbicidal activity produced by Drechslera siccans. Phytochemistry 2005, 66, 715–721. [Google Scholar] [CrossRef]
- Cao, F.; Meng, Z.H.; Mu, X.; Yue, Y.F.; Zhu, H.J. Absolute Configuration of Bioactive Azaphilones from the Marine-Derived Fungus Pleosporales sp. CF09-1. J. Nat. Prod. 2019, 82, 386–392. [Google Scholar] [CrossRef]
- Cao, F.; Meng, Z.H.; Wang, P.; Luo, D.Q.; Zhu, H.J. Dipleosporalones A and B, Dimeric Azaphilones from a Marine-Derived Pleosporales sp. Fungus. J. Nat. Prod. 2020, 83, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Bihlmeier, A.; Bourcet, E.; Arzt, S.; Muller, T.; Bräse, S.; Klopper, W. Structure Revision of Plakotenin Based on Computational Investigation of Transition States and Spectroscopic Properties. J. Am. Chem. Soc. 2012, 134, 2154–2160. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.N.; Holt, T.A.; Kutateladze, A.; Newhouse, T.R. Stereochemical revision of xylogranatin F by GIAO and DU8+ NMR calculations. Chirality 2020, 32, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Liu, Y.F.; Yue, Y.F.; Feng, L.X.; Zhu, H.J.; Cao, F. Asperienes A–D, bioactive sesquiterpenes from the marine-derived fungus Aspergillus flavus. Mar. Drugs 2019, 17, 550. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.F.; Zhang, Y.H.; Shao, C.L.; Cao, F.; Wang, C.Y. Microketides A and B, polyketides from a gorgonian-derived Microsphaeropsis sp. fungus. J. Nat. Prod. 2020, 83, 1300–1304. [Google Scholar] [CrossRef]
- Mazzeo, G.; Santoro, E.; Andolfi, A.; Cimmino, A.; Troselj, P.; Petrovic, A.G.; Superchi, S.; Evidente, A.; Berova, N. Absolute configurations of fungal and plant metabolites by chiroptical methods. ORD, ECD, and VCD studies on phyllostin, scytolide, and oxysporone. J. Nat. Prod. 2013, 76, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Mándi, A.; Kurtán, T. Applications of OR/ECD/VCD to the structure elucidation of natural products. Nat. Prod. Rep. 2019, 36, 889–918. [Google Scholar] [CrossRef]
- Zhu, A.; Yang, M.Y.; Zhang, Y.H.; Shao, C.L.; Wang, C.Y.; Hu, L.D.; Cao, F.; Zhu, H.J. Absolute configurations of 14, 15-hydroxylated prenylxanthones from a marine-derived Aspergillus sp. fungus by chiroptical methods. Sci. Rep. 2018, 8, 10621. [Google Scholar] [CrossRef] [Green Version]
- Cao, F.; Sun, T.T.; Yang, J.K.; Zhao, G.Z.; Liu, Q.A.; Hu, L.D.; Ma, Z.Y.; Zhu, H.J. The absolute configuration of anti-Vibrio citrinin dimeric derivative by VCD, ECD and NMR methods. Nat. Prod. Res. 2019, 33, 2192–2199. [Google Scholar] [CrossRef] [PubMed]
- Hussain, H.; Green, I.R.; Abbas, G.; Adekenov, S.M.; Hussain, W.; Ali, I. Protein tyrosine phosphatase 1B (PTP1B) inhibitors as potential anti-diabetes agents: Patent review (2015–2018). Expert Opin. Ther. Pat. 2019, 29, 689–702. [Google Scholar] [CrossRef]
- Kong, F.D.; Fan, P.; Zhou, L.M.; Ma, Q.Y.; Xie, Q.Y.; Zheng, H.Z.; Zheng, Z.H.; Zhang, R.S.; Yuan, J.Z.; Dai, H.F.; et al. Penerpenes A–D, four indole terpenoids with potent protein tyrosine phosphatase inhibitory activity from the marine-derived fungus Penicillium sp. KFD28. Org. Lett. 2019, 21, 4864–4867. [Google Scholar] [CrossRef]
- Jiao, W.H.; Li, J.; Zhang, M.M.; Cui, J.; Gui, Y.H.; Zhang, Y.; Li, J.Y.; Liu, K.C.; Lin, H.W. Frondoplysins A and B, Unprecedented Terpene-Alkaloid Bioconjugates from Dysidea frondosa. Org. Lett. 2019, 21, 6190–6193. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef]
- Huo, C.H.; Zheng, Z.H.; Xu, Y.; Ding, Y.B.; Zheng, H.Z.; Mu, Y.L.; Niu, Y.C.; Gao, J.; Lu, X.H. Naphthacemycins from a Streptomyces sp. as protein-tyrosine phosphatase inhibitors. J. Nat. Prod. 2020, 83, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Punthasee, P.; Laciak, A.R.; Cummings, A.H.; Ruddraraju, K.V.; Lewis, S.M.; Hillebrand, R.; Singh, H.; Tanner, J.J.; Gates, K.S. Covalent Allosteric Inactivation of Protein Tyrosine Phosphatase 1B (PTP1B) by an Inhibitor-Electrophile Conjugate. Biochemistry 2017, 56, 2051–2060. [Google Scholar] [CrossRef]
- Asante-Appiah, E.; Patel, S.; Desponts, C.; Taylor, J.M.; Lau, C.; Dufresne, C.; Therien, M.; Friesen, R.; Becker, J.W.; Leblanc, Y.; et al. Conformation-assisted inhibition of protein-tyrosine phosphatase-1B elicits inhibitor selectivity over T-cell protein-tyrosine phosphatase. J. Biol. Chem. 2006, 281, 8010–8015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koska, J.; Spassov, V.Z.; Maynard, A.J.; Yan, L.; Austin, N.; Flook, P.K.; Venkatachalam, C.M. Fully automated molecular mechanics based induced fit protein-ligand docking method. J. Chem. Inf. Model. 2008, 48, 1965–1973. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | (±)-1 Measured in CDCl3 | Drazepinone Measured in CDCl3 | ||
|---|---|---|---|---|
| δC b | δH d | δC c | δH d | |
| 2 | 103.9 | - | 104.1 | - |
| 3 | 56.8 | 3.26 brs | 56.7 | 3.26 brd (1.3) |
| 4 | 49.1 | 2.89 brq (7.2) | 49.1 | 2.90 brq (7.2) |
| 5 | 150.5 | - | 150.5 | - |
| 6 | 126.3 | 5.38 s | 126.3 | 5.38 brs |
| 7 | 164.8 a | - | 164.9 | - |
| 8 | 113.5 a | - | 113.5 | - |
| 9 | 133.6 | 7.43 s | 133.8 | 7.44 s |
| 10 | - | 11.2 brs | - | 12.4 brs |
| 11 | 162.6 a | - | 162.8 | - |
| 12 | 111.4 | - | 111.2 | - |
| 13 | 26.3 | 1.65 s | 26.3 | 1.65 s |
| 14 | 20.3 | 1.29 d (7.2) | 20.3 | 1.29 d (7.2) |
| 15 | 14.8 | 1.72 s | 14.8 | 1.72 brs |
| 16 | 133.4 | - | 133.6 | - |
| 17 | 127.6 | 7.51 d (7.8) | 127.6 | 7.51 d (7.6) |
| 18 | 128.5 | 7.38 dd (7.8,7.8) | 128.5 | 7.38 dd (7.6,7.6) |
| 19 | 127.3 | 7.30 dd (7.8,7.8) | 127.2 | 7.30 brs |
| 20 | 128.5 | 7.38 dd (7.8,7.8) | 128.5 | 7.38 dd (7.6,7.6) |
| 21 | 127.6 | 7.51 d (7.8) | 127.6 | 7.51 d (7.6) |
| Compounds | IC50 (μg/mL) | |||
|---|---|---|---|---|
| PTP1B | TCPTP | SHP2 | CD45 | |
| (+)-1 | >25.0 | >25.0 | >25.0 | >25.0 |
| (−)-1 | 1.56 | 12.5 | >25.0 | >25.0 |
| Na3VO4 | 1.56 | 3.13 | 6.25 | 6.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, F.; Pan, L.; Gao, W.; Liu, Y.; Zheng, C.; Zhang, Y. Structure Revision and Protein Tyrosine Phosphatase Inhibitory Activity of Drazepinone. Mar. Drugs 2021, 19, 714. https://doi.org/10.3390/md19120714
Cao F, Pan L, Gao W, Liu Y, Zheng C, Zhang Y. Structure Revision and Protein Tyrosine Phosphatase Inhibitory Activity of Drazepinone. Marine Drugs. 2021; 19(12):714. https://doi.org/10.3390/md19120714
Chicago/Turabian StyleCao, Fei, Li Pan, Wenbin Gao, Yunfeng Liu, Caijuan Zheng, and Yahui Zhang. 2021. "Structure Revision and Protein Tyrosine Phosphatase Inhibitory Activity of Drazepinone" Marine Drugs 19, no. 12: 714. https://doi.org/10.3390/md19120714