Novel Macrolactams from a Deep-Sea-Derived Streptomyces Species

 and
and
Abstract
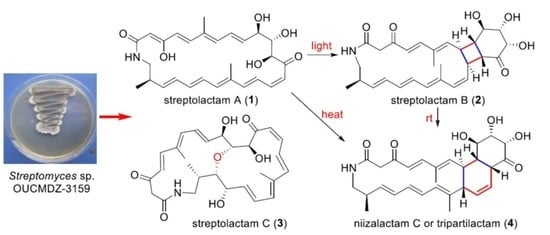
:
1. Introduction
2. Results and Discussion
2.1. Structural Elucidation
2.2. The Bioactivities of Compounds 1–4 from Streptomyces sp. OUCMDZ-3159
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Collection and Phylogenetic Analysis
3.3. Cultivation and Extraction
3.4. Purification
3.5. Preparation of Compounds 1a and 3a
3.6. Preparation of Compound 1b
3.7. Preparation of S-MTPA Ester (1ba) and R-MTPA Ester (1bb) of Compound 1b
3.8. Characterization of the Compounds
3.9. X-Ray Crystallographic Analysis
3.10. Chemical Interconversion of Compounds 1,2 and 4
3.11. Cytotoxicity Assay
3.12. Antimicrobial Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Skellam, E.J.; Stewart, A.K.; Strangman, W.K.; Wright, J.L.C. Identification of micromonolactam, a new polyene macrocyclic lactam from two marine Micromonospora strains using chemical and molecular methods: Clarification of the biosynthetic pathway from a glutamate starter unit. J. Antibiot. 2013, 66, 431–441. [Google Scholar]
- Sugiyama, R.; Nishimura, S.; Matsumori, N.; Tsunematsu, Y.; Hattori, A.; Kakeya, H. Structure and biological activity of 8-deoxyheronamide C from a marine-derived Streptomyces sp.: Heronamides target saturated hydrocarbon chains in lipid membranes. J. Am. Chem. Soc. 2014, 136, 5209–5212. [Google Scholar] [PubMed]
- Schulze, C.J.; Donia, M.S.; Siqueira-Neto, J.L.; Ray, D.; Raskatov, J.A.; Green, R.E.; McKerrow, J.H.; Fischbach, M.A.; Linington, R.G. Genome-directed lead discovery: Biosynthesis, structure elucidation, and biological evaluation of two families of polyene macrolactams against Trypanosoma brucei. ACS Chem. Biol. 2015, 10, 2373–2381. [Google Scholar] [PubMed]
- Genilloud, O. Actinomycetes: Still a source of novel antibiotics. Nat. Prod. Rep. 2017, 34, 1203–1232. [Google Scholar]
- Hoshino, S.; Okada, M.; Awakawa, T.; Asamizu, S.; Onaka, H.; Abe, I. Mycolic acid containing bacterium stimulates tandem cyclization of polyene macrolactam in a lake sediment derived rare actinomycete. Org. Lett. 2017, 19, 4992–4995. [Google Scholar]
- Shin, Y.H.; Beom, J.Y.; Chung, B.; Shin, Y.; Byun, W.S.; Moon, K.; Bae, M.; Lee, S.K.; Oh, K.B.; Shin, J.; et al. Bombyxamycins A and B, cytotoxic macrocyclic lactams from an intestinal bacterium of the silkworm Bombyx mori. Org. Lett. 2019, 21, 1804–1808. [Google Scholar]
- Beemelmanns, C.; Ramadhar, T.R.; Kim, K.H.; Klassen, J.L.; Cao, S.; Wyche, T.P.; Hou, Y.; Poulsen, M.; Bugni, T.S.; Currie, C.R.; et al. Macrotermycins A–D, glycosylated macrolactams from a termite-associated Amycolatopsis sp. M39. Org. Lett. 2017, 19, 1000–1003. [Google Scholar]
- Nogawa, T.; Okano, A.; Takahashi, S.; Uramoto, M.; Konno, H.; Saito, T.; Osada, H. Verticilactam, a new macrolactam isolated from a microbial metabolite fraction library. Org. Lett. 2010, 12, 4564–4567. [Google Scholar]
- Oh, D.C.; Poulsen, M.; Currie, C.R.; Clardy, J. Sceliphrolactam, a polyene macrocyclic lactam from a wasp-associated Streptomyces sp. Org. Lett. 2011, 13, 752–755. [Google Scholar]
- Park, S.H.; Moon, K.; Bang, H.S.; Kim, S.H.; Kim, D.G.; Oh, K.B.; Shin, J.; Oh, D.C. Tripartilactam, a cyclobutane-bearing tricyclic lactam from a Streptomyces sp. in a dung beetle’s brood ball. Org. Lett. 2012, 14, 1258–1261. [Google Scholar]
- Hoshino, S.; Okada, M.; Wakimoto, T.; Zhang, H.P.; Hayashi, F.; Onaka, H.; Abe, I. Niizalactams A–C, multicyclic macrolactams isolated from combined culture of Streptomyces with mycolic acid-containing bacterium. J. Nat. Prod. 2015, 78, 3011–3017. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Kim, E.; Lee, J.; Shin, J.; Yoon, Y.J.; Oh, D.C. Structure revision and the biosynthetic pathway of tripartilactam. J. Nat. Prod. 2020, 83, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Liu, P.; Li, X.; Wang, Y.; Wang, S.; Hong, K.; Zhu, W. Cyclic bipyridine glycosides from the marine-derived actinomycete Actinoalloteichus cyanogriseus WH1-2216-6. Org. Lett. 2011, 13, 5948–5951. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Zhu, Y.; Mei, X.; Wang, Y.; Jia, H.; Zhang, C.; Zhu, W. Acyclic congeners from Actinoalloteichus cyanogriseus provide insights into cyclic bipyridine glycoside formation. Org. Lett. 2014, 16, 4264–4267. [Google Scholar] [CrossRef]
- Shen, J.; Fan, Y.; Zhu, G.; Chen, H.; Zhu, W.; Fu, P. Polycyclic macrolactams generated via intramolecular diels–alder reactions from an Antarctic Streptomyces species. Org. Lett. 2019, 21, 4816–4820. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wang, J.; Chen, H.; Wang, Y.; Zhu, W.; Fu, P. Cyclamenols E and F, two diastereoisomeric bicyclic macrolactams with a cyclopentane moiety from an Antarctic Streptomyces species. Org. Chem. Front. 2020, 7, 310–317. [Google Scholar] [CrossRef]
- Seco, J.M.; Quiñoá, E.; Riguera, R. A practical guide for the assignment of the absolute configuration of alcohols, amines and carboxylic acids by NMR. Tetrahedron Asymmetry 2001, 12, 2915–2925. [Google Scholar] [CrossRef]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon-proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Xiong, T.; Chen, X.; Wei, H.; Xiao, H. Influence of PJ34 on the genotoxicity induced by melphalan in human multiple myeloma cells. Arch. Med. Sci. 2015, 11, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Zaika, L.L.J. Spices and herbs: Their antimicrobial activity and its determination. J. Food Safety 1988, 9, 97–118. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 (in DMSO-d6) | 2 (in Pyridine-d5) b | 3 (in DMSO-d6) | |||
|---|---|---|---|---|---|---|
| δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | |
| 1 | 172.4, C | 166.3, C | 166.8, C | |||
| 2 | 93.7, CH | 4.99, s | 50.1, CH2 | 3.86, d (14.8); 3.65, d (14.8) | 50.4, CH2 | 3.91, overlapped; 3.43, d (17.9) |
| 3 | 165.8, C | 194.8, C | 193.3, C | |||
| 4 | 121.6 a, CH | 5.84, d (15.3) | 123.4 a, CH | 6.27, d (15.0) | 123.0, CH | 6.11, d (15.4) |
| 5 | 138.2, CH | 6.54, d (15.3) | 149.4, CH | 7.28, d (15.0) | 146.3, CH | 6.74, d (15.4) |
| 6 | 133.1, C | 134.1, C | 133.2, C | |||
| 7 | 135.9, CH | 5.86, d (10.3) | 146.2, CH | 6.07, d (10.6) | 141.0, CH | 6.04, d (11.2) |
| 8 | 128.4, CH | 6.37, dd (13.8, 12.9) | 42.6, CH | 4.59, “t” like (10.2) | 129.7, CH | 6.32, dd (14.5, 11.6) |
| 9 | 136.6, CH | 5.43, dd (13.8, 7.8) | 50.5, CH | 3.52, “t” like (10.6) | 139.0, CH | 5.45, dd (14.6, 9.2) |
| 10 | 70.0, CH | 4.04, t, (7.8) | 70.4, CH | 4.40, m | 70.0, CH | 4.00, t (9.1) |
| 11 | 74.3 a, CH | 3.79, d, (8.3) | 81.8, CH | 5.17, m | 73.3, CH | 3.82, overlapped |
| 12 | 79.6, CH | 4.26, brs | 76.6, CH | 5.62, m | 79.7, CH | 4.33, brs |
| 13 | 199.1, C | 213.1, C | 198.6 a, C | |||
| 14 | 119.9, CH | 6.24, overlapped | 51.8, CH | 3.22, m | 120.8, CH | 6.13, d (11.5) |
| 15 | 143.5, CH | 6.68, t (11.2) | 40.6, CH | 4.31, m | 143.8, CH | 6.67, t (11.4) |
| 16 | 125.6, CH | 7.39, t (13.4) | 129.6, CH | 5.64, m | 126.6, CH | 7.39, dd (15.0, 11.8) |
| 17 | 146.4, CH | 6.64, d (15.0) | 137.9, CH | 6.21, d (11.4) | 146.9, CH | 6.61, d (15.3) |
| 18 | 134.6 a, C | 136.3, C | 136.2, C | |||
| 19 | 135.8, CH | 6.24, overlapped | 132.9, CH | 6.03, d (11.0) | 135.1, CH | 6.14, d (11.6) |
| 20 | 135.3 a, CH | 6.18, overlapped | 128.0, CH | 6.10, dd (14.0, 11.0) | 129.2, CH | 6.38, dd (14.4, 11.6) |
| 21 | 127.4, CH | 6.20, overlapped | 133.7, CH | 6.30, dd (14.0, 11.0) | 137.6, CH | 5.45, dd (14.6, 9.2) |
| 22 | 131.4, CH | 5.81, t (15.2, 9.2) | 133.2, CH | 5.96, dd (15.0, 11.0) | 67.9, CH | 3.93, overlapped |
| 23 | 137.4, CH | 5.36, m | 137.7, CH | 5.42, overlapped | 82.4, CH | 3.46, dd (8.5, 7.5) |
| 24 | 39.5 a, CH | 2.20, brs | 38.7, CH | 2.59, m | 38.5, CH | 1.83, m |
| 25 | 43.7, CH2 | 3.00, m; 3.06, m | 46.3, CH2 | 3.02, m; 3.61, m | 50.6, CH2 | 2.78 d (11.2, 10.9); 3.83, overlapped |
| 26 | 12.0, CH3 | 1.80, s | 13.0, CH3 | 1.86, s | 12.4, CH3 | 1.77, s |
| 27 | 12.0, CH3 | 1.60, s | 16.0, CH3 | 1.58, s | 12.5, CH3 | 1.61, s |
| 28 | 16.2, CH3 | 1.00, d (6.5) | 18.1, CH3 | 0.84, d (5.3) | 14.8, CH3 | 0.99, d (6.4) |
| -NH | 7.73, dd (5.4, 6.1) | 8.34, brs | ||||
| 3-OH | 13.69, s | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Wang, D.; Zhang, R.; Wang, Y.; Kong, F.; Fu, P.; Zhu, W. Novel Macrolactams from a Deep-Sea-Derived Streptomyces Species. Mar. Drugs 2021, 19, 13. https://doi.org/10.3390/md19010013
Wang P, Wang D, Zhang R, Wang Y, Kong F, Fu P, Zhu W. Novel Macrolactams from a Deep-Sea-Derived Streptomyces Species. Marine Drugs. 2021; 19(1):13. https://doi.org/10.3390/md19010013
Chicago/Turabian StyleWang, Pei, Dongyang Wang, Rongxin Zhang, Yi Wang, Fandong Kong, Peng Fu, and Weiming Zhu. 2021. "Novel Macrolactams from a Deep-Sea-Derived Streptomyces Species" Marine Drugs 19, no. 1: 13. https://doi.org/10.3390/md19010013