Fucosterol Suppresses the Progression of Human Ovarian Cancer by Inducing Mitochondrial Dysfunction and Endoplasmic Reticulum Stress


Abstract
:1. Introduction
2. Results
2.1. Fucosterol Suppressed Cell Growth and Induced Apoptosis in Ovarian Cancer Cells
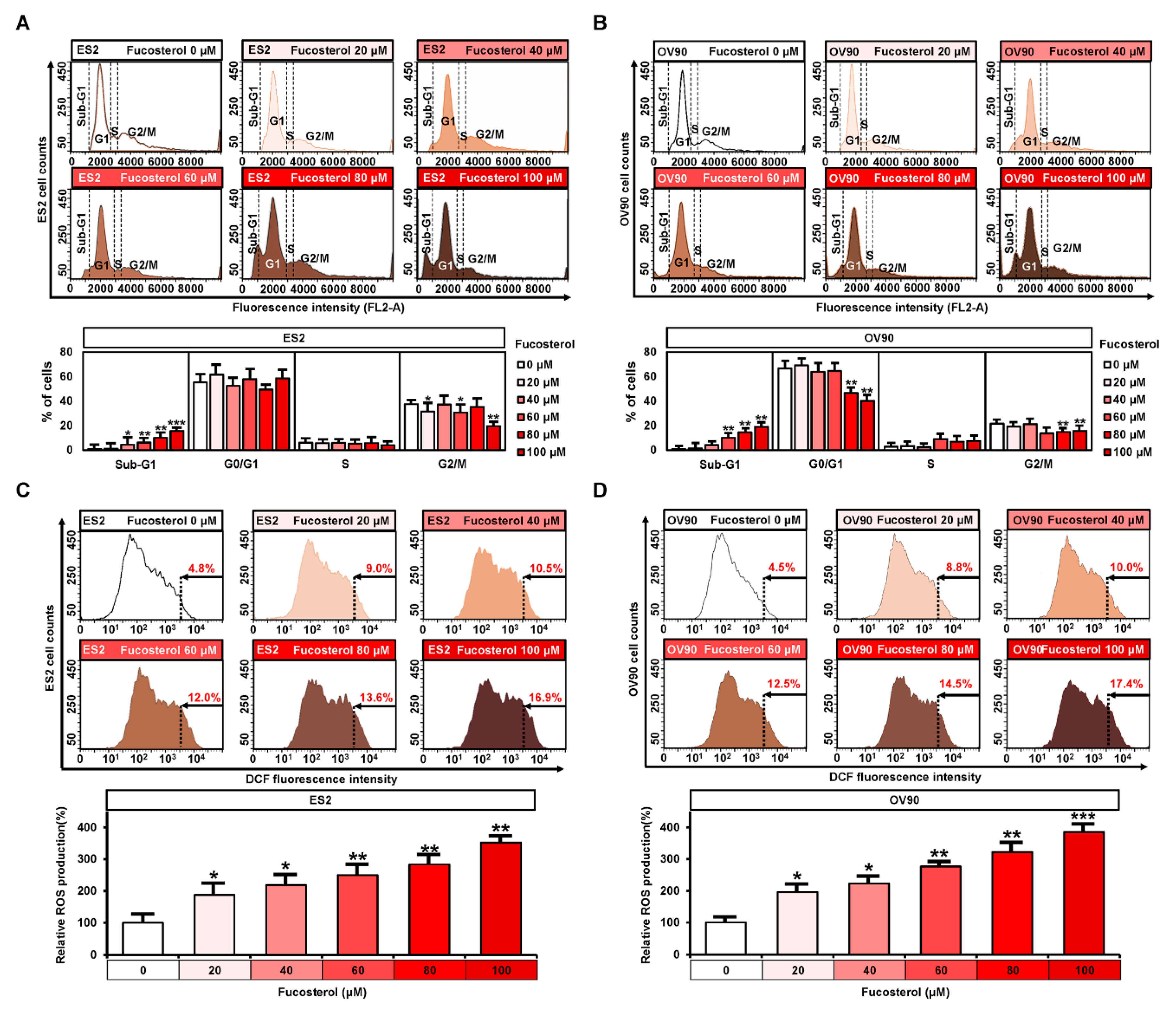
2.2. Regulation of Fucosterol on Cell Cycle Progression and ROS Production
2.3. Effect of Fucosterol on the Concentration of Calcium Ions, Mitochondrial Membrane Potential (MMP), and Apoptotic Proteins in Both Cell Lines
2.4. Fucosterol Inactivated PI3K/MAPK Signal Transduction in Ovarian Cancer Cells
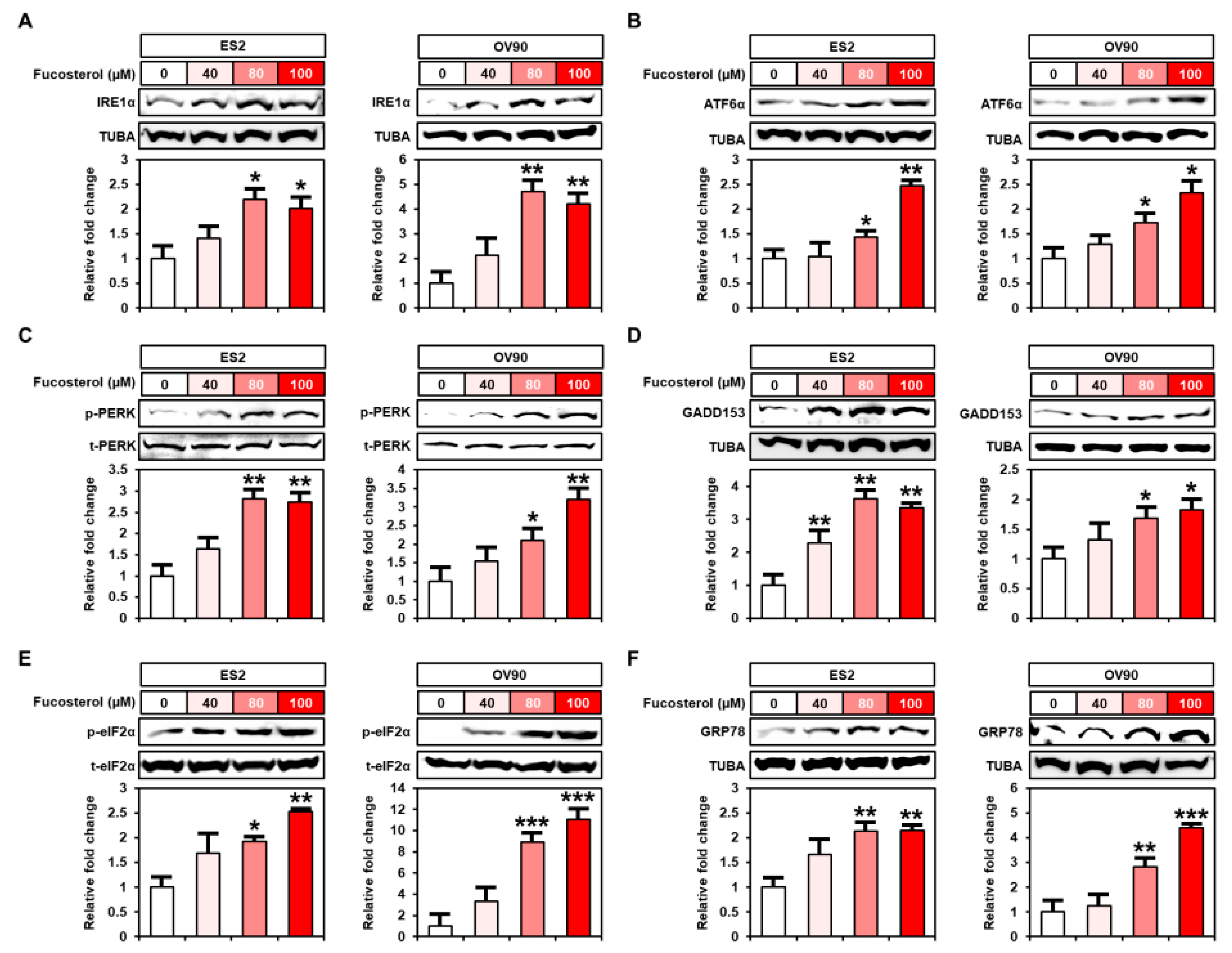
2.5. Enhanced Endoplasmic Reticulum (ER) Stress Caused by Fucosterol in Human Ovarian Cancer Cells
2.6. Synergistic Effects of Fucosterol and Cisplatin or Paclitaxel on Apoptotic Signaling Pathways and Anti-Angiogenesis Effects
2.7. Effects of Fucosterol on Cytotoxicity, Tumorigenesis, and Angiogenesis In Vivo
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Proliferation Assay
4.4. Immunofluorescence Microscopy
4.5. Annexin V and PI Staining
4.6. Cell-Cycle Assay
4.7. Determination of Cellular ROS
4.8. Measurement of Concentrations of Cytosol Ca2+
4.9. Measurement of Mitochondrial Ca2+ Concentration
4.10. JC-1 Mitochondrial Membrane Potential (MMP) Assay
4.11. Western Blot Analysis
4.12. RNA Isolation
4.13. Quantitative PCR Analysis
4.14. Toxicity and Angiogenesis Analysis In Vivo
4.15. Xenografts
4.16. Statistical Analyses
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, K. Treatment for recurrent ovarian cancer-at first relapse. J. Oncol. 2010, 2010, 497429. [Google Scholar] [CrossRef] [PubMed]
- McGuire, W.P.; Hoskins, W.J.; Brady, M.F.; Kucera, P.R.; Partridge, E.E.; Look, K.Y.; Clarke-Pearson, D.L.; Davidson, M. Cyclophosphamide and cisplatin versus paclitaxel and cisplatin: A phase III randomized trial in patients with suboptimal stage III/IV ovarian cancer (from the Gynecologic Oncology Group). Semin. Oncol. 1996, 23, 40–47. [Google Scholar] [PubMed]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, A.R.; Tiwari, U.; Rajauria, G. Seaweed nutraceuticals and their therapeutic role in disease prevention. Food Sci. Hum. Wellness 2019, 8, 252–263. [Google Scholar] [CrossRef]
- Abdul, Q.A.; Choi, R.J.; Jung, H.A.; Choi, J.S. Health benefit of fucosterol from marine algae: A review. J. Sci. Food Agric. 2016, 96, 1856–1866. [Google Scholar] [CrossRef]
- Zhen, X.H.; Quan, Y.C.; Jiang, H.Y.; Wen, Z.S.; Qu, Y.L.; Guan, L.P. Fucosterol, a sterol extracted from Sargassum fusiforme, shows antidepressant and anticonvulsant effects. Eur. J. Pharmacol. 2015, 768, 131–138. [Google Scholar] [CrossRef]
- Sheu, J.H.; Wang, G.H.; Sung, P.J.; Chiu, Y.H.; Duh, C.Y. Cytotoxic sterols from the formosan brown alga Turbinaria ornata. Planta Med. 1997, 63, 571–572. [Google Scholar] [CrossRef]
- Khanavi, M.; Gheidarloo, R.; Sadati, N.; Ardekani, M.R.; Nabavi, S.M.; Tavajohi, S.; Ostad, S.N. Cytotoxicity of fucosterol containing fraction of marine algae against breast and colon carcinoma cell line. Pharmacogn. Mag. 2012, 8, 60–64. [Google Scholar] [CrossRef]
- Pacheco, B.S.; Dos Santos, M.A.Z.; Schultze, E.; Martins, R.M.; Lund, R.G.; Seixas, F.K.; Colepicolo, P.; Collares, T.; Paula, F.R.; De Pereira, C.M.P. Cytotoxic Activity of Fatty Acids From Antarctic Macroalgae on the Growth of Human Breast Cancer Cells. Front. Bioeng. Biotechnol. 2018, 6, 185. [Google Scholar] [CrossRef] [PubMed]
- Sheu, J.H.; Wang, G.H.; Sung, P.J.; Duh, C.Y. New cytotoxic oxygenated fucosterols from the brown alga Turbinaria conoides. J. Nat. Prod. 1999, 62, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Li, J.; Chen, A.; Li, Y.; Xia, M.; Guo, P.; Yao, S.; Chen, S. Fucosterol exhibits selective antitumor anticancer activity against HeLa human cervical cell line by inducing mitochondrial mediated apoptosis, cell cycle migration inhibition and downregulation of m-TOR/PI3K/Akt signalling pathway. Oncol. Lett. 2018, 15, 3458–3463. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, Z.; Liu, X.; Teng, H.; Zhang, C.; Hou, L.; Zou, X. Anti-metastasis effect of fucoidan from Undaria pinnatifida sporophylls in mouse hepatocarcinoma Hca-F cells. PLoS ONE 2014, 9, e106071. [Google Scholar] [CrossRef] [Green Version]
- Lichota, A.; Gwozdzinski, K. Anticancer Activity of Natural Compounds from Plant and Marine Environment. Int. J. Mol. Sci. 2018, 19, 3533. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Kim, M.K.; Lee, M.; Kwon, B.S.; Suh, D.H.; Song, Y.S. Effect of Red Ginseng on Genotoxicity and Health-Related Quality of Life after Adjuvant Chemotherapy in Patients with Epithelial Ovarian Cancer: A Randomized, Double Blind, Placebo-Controlled Trial. Nutrients 2017, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Yunos, N.M.; Beale, P.; Yu, J.Q.; Strain, D.; Huq, F. Studies on combinations of platinum with paclitaxel and colchicine in ovarian cancer cell lines. Anticancer Res. 2010, 30, 4025–4037. [Google Scholar]
- Tang, H.F.; Yang-Hua, Y.; Yao, X.S.; Xu, Q.Z.; Zhang, S.Y.; Lin, H.W. Bioactive steroids from the brown alga Sargassum carpophyllum. J. Asian Nat. Prod. Res. 2002, 4, 95–101. [Google Scholar] [CrossRef]
- Byju, K.; Anuradha, V.; Vasundhara, G.; Nair, S.M.; Kumar, N.C. In vitro and in silico studies on the anticancer and apoptosis-inducing activities of the sterols identified from the soft coral, subergorgia reticulata. Pharmacogn. Mag. 2014, 10, S65–S71. [Google Scholar] [CrossRef] [Green Version]
- Suttiarporn, P.; Chumpolsri, W.; Mahatheeranont, S.; Luangkamin, S.; Teepsawang, S.; Leardkamolkarn, V. Structures of phytosterols and triterpenoids with potential anti-cancer activity in bran of black non-glutinous rice. Nutrients 2015, 7, 1672–1687. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.B.; Ji, C.F.; Yue, L. Study on human promyelocytic leukemia HL-60 cells apoptosis induced by fucosterol. Biomed. Mater. Eng. 2014, 24, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xie, J.; Li, J.; Liu, F.; Wu, X.; Wang, Z. Cisplatin suppresses the growth and proliferation of breast and cervical cancer cell lines by inhibiting integrin beta5-mediated glycolysis. Am. J. Cancer Res. 2016, 6, 1108–1117. [Google Scholar] [PubMed]
- Hanna, R.K.; Zhou, C.X.; Malloy, K.M.; Sun, L.; Zhong, Y.; Gehrig, P.A.; Bae-Jump, V.L. Metformin potentiates the effects of paclitaxel in endometrial cancer cells through inhibition of cell proliferation and modulation of the mTOR pathway. Gynecol. Oncol. 2012, 125, 458–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malkas, L.H.; Herbert, B.S.; Abdel-Aziz, W.; Dobrolecki, L.E.; Liu, Y.; Agarwal, B.; Hoelz, D.; Badve, S.; Schnaper, L.; Arnold, R.J.; et al. A cancer-associated PCNA expressed in breast cancer has implications as a potential biomarker. Proc. Natl. Acad. Sci. USA 2006, 103, 19472–19477. [Google Scholar] [CrossRef] [Green Version]
- Murad, H.; Hawat, M.; Ekhtiar, A.; AlJapawe, A.; Abbas, A.; Darwish, H.; Sbenati, O.; Ghannam, A. Induction of G1-phase cell cycle arrest and apoptosis pathway in MDA-MB-231 human breast cancer cells by sulfated polysaccharide extracted from Laurencia papillosa. Cancer Cell Int. 2016, 16, 39. [Google Scholar] [CrossRef] [Green Version]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin Induces a Mitochondrial-ROS Response That Contributes to Cytotoxicity Depending on Mitochondrial Redox Status and Bioenergetic Functions. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [Green Version]
- Azimi, I.; Roberts-Thomson, S.J.; Monteith, G.R. Calcium influx pathways in breast cancer: Opportunities for pharmacological intervention. Br. J. Pharmacol. 2014, 171, 945–960. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.F.; Hu, Y.C.; Kang, B.H.; Tseng, Y.K.; Wu, P.C.; Liang, C.C.; Hou, Y.Y.; Fu, T.Y.; Liou, H.H.; Hsieh, I.C.; et al. Expression levels of cleaved caspase-3 and caspase-3 in tumorigenesis and prognosis of oral tongue squamous cell carcinoma. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.X.; Xu, G.P.; Yeung, S.C.J. Cytochrome c release is upstream to activation of caspase-9, caspase-8, and caspase-3 in the enhanced apoptosis of anaplastic thyroid cancer cells induced by manumycin and paclitaxel. J. Clin. Endocr. Metab. 2001, 86, 4731–4740. [Google Scholar] [CrossRef]
- Zhou, H.; Zhao, H.; Liu, H.; Xu, X.; Dong, X.; Zhao, E. Influence of carboplatin on the proliferation and apoptosis of ovarian cancer cells through mTOR/p70s6k signaling pathway. J. BUON 2018, 23, 1732–1738. [Google Scholar] [PubMed]
- Ventura, A.P.; Radhakrishnan, S.; Green, A.; Rajaram, S.K.; Allen, A.N.; O’Briant, K.; Schummer, M.; Karlan, B.; Urban, N.; Tewari, M.; et al. Activation of the MEK-S6 pathway in high-grade ovarian cancers. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 499–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.I.; Park, S.H.; Lee, H.J.; Lee, D.W.; Lee, H.N. Inhibition of Phospho-S6 Kinase, a Protein Involved in the Compensatory Adaptive Response, Increases the Efficacy of Paclitaxel in Reducing the Viability of Matrix-Attached Ovarian Cancer Cells. PLoS ONE 2016, 11, e0155052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Wang, J.; Xiao, H.; Xiao, C.; Wang, Y.; Liu, X. Isoorientin induces apoptosis through mitochondrial dysfunction and inhibition of PI3K/Akt signaling pathway in HepG2 cancer cells. Toxicol. Appl. Pharmacol. 2012, 265, 83–92. [Google Scholar] [CrossRef]
- Larman, M.; Lucy, J. The endoplasmic reticulum: Structure and function. Mol. Membr. Biol. 1999, 16, 313–316. [Google Scholar]
- Malhotra, J.D.; Kaufman, R.J. ER stress and its functional link to mitochondria: Role in cell survival and death. Cold Spring Harb. Perspect. Biol. 2011, 3, a004424. [Google Scholar] [CrossRef]
- Bhatta, M.; Chatpar, K.; Hu, Z.; Wang, J.J.; Zhang, S.X. Reduction of Endoplasmic Reticulum Stress Improves Angiogenic Progenitor Cell function in a Mouse Model of Type 1 Diabetes. Cell Death Dis. 2018, 9, 467. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Qiu, T.; Wu, J.; Yang, Y.; Wright, G.D.; Wu, M.; Ge, R. Extracellular anti-angiogenic proteins augment an endosomal protein trafficking pathway to reach mitochondria and execute apoptosis in HUVECs. Cell Death Differ. 2018, 25, 1905–1920. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.B.; Zhi, H.; Zhang, X.Y.; Liang, J.W.; He, J.; Su, C.; Xia, W.H.; Zhang, G.X.; Tao, J. Mitochondrial dysfunction-mediated decline in angiogenic capacity of endothelial progenitor cells is associated with capillary rarefaction in patients with hypertension via downregulation of CXCR4/JAK2/SIRT5 signaling. Ebiomedicine 2019, 42, 64–75. [Google Scholar] [CrossRef] [Green Version]
- Gbel, J.; Engelhardt, E.; Pelzer, P.; Sakthivelu, V.; Jahn, H.M.; Jevtic, M.; Folz-Donahue, K.; Kukat, C.; Schauss, A.; Frese, C.K.; et al. Mitochondria-Endoplasmic Reticulum Contacts in Reactive Astrocytes Promote Vascular Remodeling. Cell Metab. 2020, 31, 791–808. [Google Scholar] [CrossRef]
- Yeung, T.L.; Leung, C.S.; Yip, K.P.; Au Yeung, C.L.; Wong, S.T.; Mok, S.C. Cellular and molecular processes in ovarian cancer metastasis. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C444–C456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.A.; Almeida, T.; Lima, B.; Rocha, E. Cytotoxic activity of the seaweed compound fucosterol, alone and in combination with 5-fluorouracil, in colon cells using 2D and 3D culturing. J. Toxicol. Environ. Health A 2019, 82, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.; An, Y.; Yang, C.; Bazer, F.W.; Song, G. Chrysophanol induces cell death and inhibits invasiveness via mitochondrial calcium overload in ovarian cancer cells. J. Cell Biochem. 2018, 119, 10216–10227. [Google Scholar] [CrossRef]
- Lee, J.Y.; Talhi, O.; Jang, D.; Cerella, C.; Gaigneaux, A.; Kim, K.W.; Lee, J.W.; Dicato, M.; Bachari, K.; Han, B.W.; et al. Cytostatic hydroxycoumarin OT52 induces ER/Golgi stress and STAT3 inhibition triggering non-canonical cell death and synergy with BH3 mimetics in lung cancer. Cancer Lett. 2018, 416, 94–108. [Google Scholar] [CrossRef]
- Lian, I.A.; Loset, M.; Mundal, S.B.; Fenstad, M.H.; Johnson, M.P.; Eide, I.P.; Bjorge, L.; Freed, K.A.; Moses, E.K.; Austgulen, R. Increased endoplasmic reticulum stress in decidual tissue from pregnancies complicated by fetal growth restriction with and without pre-eclampsia. Placenta 2011, 32, 823–829. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies | Dilution | Supplier | Catalog Number |
|---|---|---|---|
| Phospho-Cyclin D1 (Thr286) | 1:1000 | Cell Signaling | 3300 |
| Cyclin D1 | 1:1000 | Cell Signaling | 2922 |
| PCNA | 1:100 | Santa Cruz | sc-56 |
| Phospho-AKT (SER473) | 1:1000 | Cell Signaling | 4060 |
| AKT | 1:1000 | Cell Signaling | 9272 |
| Phospho-P70S6K (Thr421/Ser424) | 1:1000 | Cell Signaling | 9204 |
| P70S6K | 1:1000 | Cell Signaling | 9202 |
| Phospho-S6 (Ser235/236) | 1:1000 | Cell Signaling | 2211 |
| S6 | 1:1000 | Cell Signaling | 2217 |
| Phospho-ERK1/2(Thr202/Tyr204) | 1:1000 | Cell Signaling | 9101 |
| ERK1/2 | 1:1000 | Cell Signaling | 4695 |
| Phospho-JNK (Thr183/Tyr185) | 1:1000 | Cell Signaling | 4668 |
| JNK | 1:1000 | Cell Signaling | 9252 |
| Phospho-P38 (Thr180/Tyr182) | 1:1000 | Cell Signaling | 4511 |
| P38 | 1:1000 | Cell Signaling | 9212 |
| IRE1α | 1:1000 | Cell Signaling | 3294 |
| ATF6α | 1:1000 | Santa Cruz | sc-166659 |
| Phospho-PERK (Thr981) | 1:1000 | Santa Cruz | sc-32577 |
| PERK | 1:1000 | Santa Cruz | sc-13073 |
| GADD153 | 1:1000 | Santa Cruz | sc-7351 |
| Phospho-eIF2α (Ser51) | 1:1000 | Cell Signaling | 3398 |
| eIF2α | 1:1000 | Cell Signaling | 5324 |
| GRP78 | 1:1000 | Santa Cruz | sc-13968 |
| Cleaved caspase-3 | 1:1000 | Cell Signaling | 9664 |
| Cleaved caspase-9 | 1:1000 | Cell Signaling | 9501 |
| Cytochrome c | 1:1000 | Cell Signaling | 11940 |
| TUBA | 1:1000 | Santa Cruz | sc-5286 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, H.; Lee, J.-Y.; Song, G.; Lim, W. Fucosterol Suppresses the Progression of Human Ovarian Cancer by Inducing Mitochondrial Dysfunction and Endoplasmic Reticulum Stress. Mar. Drugs 2020, 18, 261. https://doi.org/10.3390/md18050261
Bae H, Lee J-Y, Song G, Lim W. Fucosterol Suppresses the Progression of Human Ovarian Cancer by Inducing Mitochondrial Dysfunction and Endoplasmic Reticulum Stress. Marine Drugs. 2020; 18(5):261. https://doi.org/10.3390/md18050261
Chicago/Turabian StyleBae, Hyocheol, Jin-Young Lee, Gwonhwa Song, and Whasun Lim. 2020. "Fucosterol Suppresses the Progression of Human Ovarian Cancer by Inducing Mitochondrial Dysfunction and Endoplasmic Reticulum Stress" Marine Drugs 18, no. 5: 261. https://doi.org/10.3390/md18050261