Terpenoids from the Deep-Sea-Derived Fungus Penicillium thomii YPGA3 and Their Bioactivities

Abstract
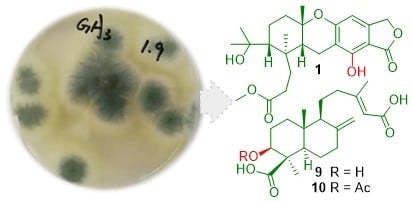
:
1. Introduction
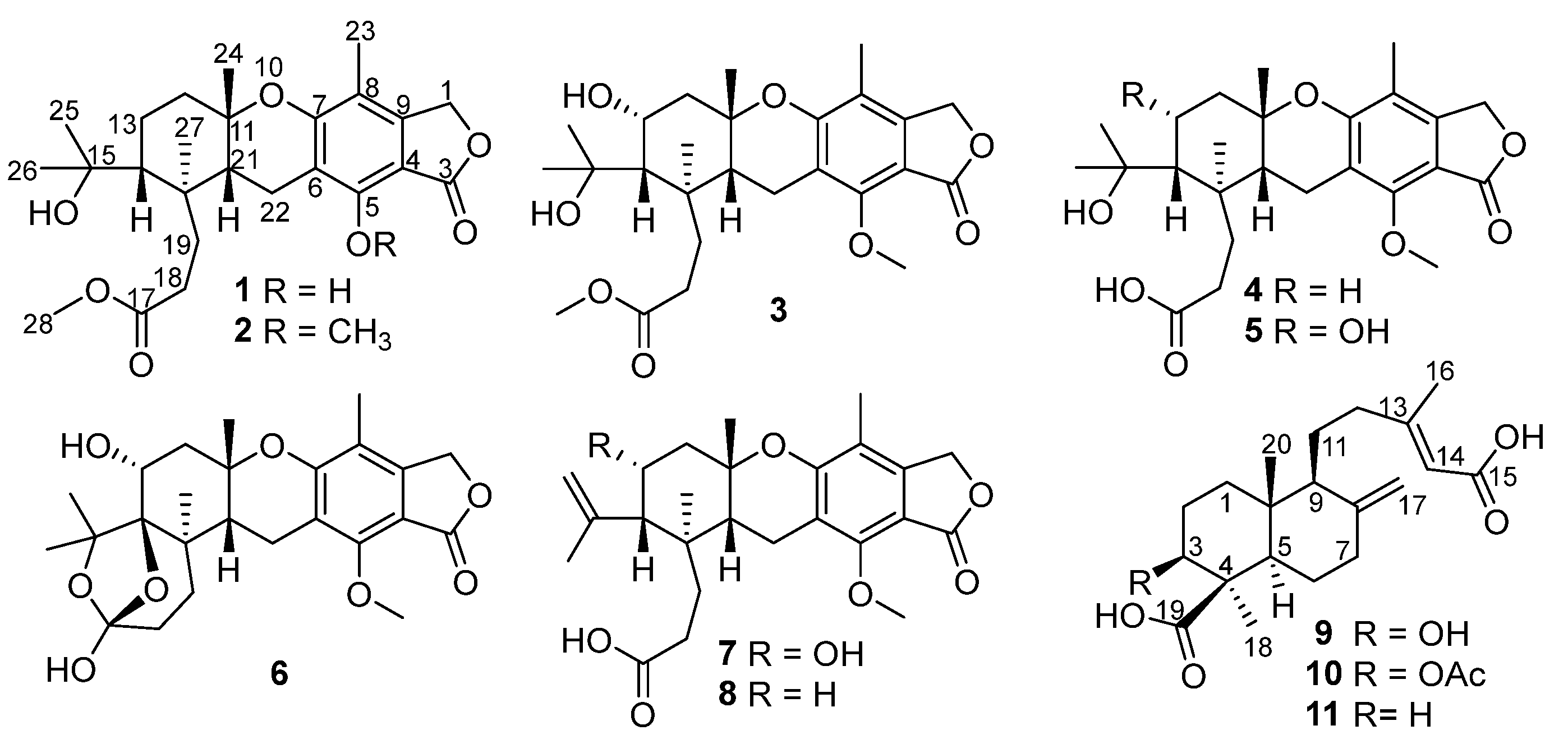
2. Results
3. Experimental Section
3.1. General Experimental Procedure
3.2. Fungal Strain and Identification
3.3. Fermentation
3.4. Extraction and Isolation
3.5. α-Glucosidase Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- de Jesus, A.E.; Horak, R.M.; Steyn, P.S.; Vleggaar, R. Biosynthesis of austalide D, a meroterpenoid mycotoxin from Aspergillus ustus. J. Chem. Soc. Chem. Commun. 1983, 13, 716–718. [Google Scholar] [CrossRef]
- Horak, R.M.; Steyn, P.S.; Van Rooyen, P.H.; Vleggaar, R.; Rabie, C.J. Structures of the austalides A–E, five noval toxic metabolites from Aspergillus ustus. J. Chem. Soc. Chem. Commun. 1981, 24, 1265–1267. [Google Scholar] [CrossRef]
- Horak, R.M.; Steyn, P.S.; Vleggaar, R.; Rabie, C.J. Metabolites of Aspergillus ustus. Part 1. A of the heteronuclear selective population inversion (SPI) n.m.r. technique to the structure elucidation of the austalides A–F, novel ortho ester meroterpenoids. J. Chem. Soc.-Perkin Trans. 1 1985, 345–356. [Google Scholar] [CrossRef]
- Horak, R.M.; Steyn, P.S.; Vleggaar, R. Metabolites of Aspergillus ustus. Part 2. Stereoelectronic control in the acid-catalysed hydrolysis of the ortho ester moiety in austalides A–F. J. Chem. Soc.-Perkin Trans. 1 1985, 357–361. [Google Scholar] [CrossRef]
- Horak, R.M.; Steyn, P.S.; Vleggaar, R.; Rabie, C.J. Metabolites of Aspergillus ustus. Part 3. Structure elucidation of austalides G–L. J. Chem. Soc.-Perkin Trans. 1 1985, 363–367. [Google Scholar] [CrossRef]
- Zhou, Y.; Mándi, A.; Debbab, A.; Wray, V.; Schulz, B.; Müller, W.E.G.; Lin, W.; Proksch, P.; Kurtán, T.; Aly, A.H. New austalides from the sponge-associated fungus Aspergillus sp. Eur. J. Org. Chem. 2011, 2011, 6009–6019. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Debbab, A.; Wray, V.; Lin, W.; Schulz, B.; Trepos, R.; Pile, C.; Hellio, C.; Proksch, P.; Aly, A.H. Marine bacterial inhibitors from the sponge-derived fungus Aspergillus sp. Tetrahedron Lett. 2014, 55, 2789–2792. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Zhang, X.; Wang, W.; Zhu, T.; Gu, Q.; Li, D. Austalides S–U, New meroterpenoids from the sponge-derived fungus Aspergillus aureolatus HDN14-107. Mar. Drugs 2016, 14, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuravleva, O.I.; Sobolevskaya, M.P.; Leshchenko, E.V.; Kirichuk, N.N.; Denisenko, V.A.; Dmitrenok, P.S.; Dyshlovoy, S.A.; Zakharenko, A.M.; Kim, N.Y.; Afiyatullov, S.S. Meroterpenoids from the alga-derived fungi Penicillium thomii Maire and Penicillium lividum Westling. J. Nat. Prod. 2014, 77, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Antipova, T.V.; Zaitsev, K.V.; Oprunenko, Y.F.; Zherebker, A.Y.; Rystsov, G.K.; Zemskova, M.Y.; Zhelifonova, V.P.; Ivanushkina, N.E.; Kozlovsky, A.G. Austalides V and W, new meroterpenoids from the fungus Aspergillus ustus and their antitumor activities. Bioorg. Med. Chem. Lett. 2019, 29, 126708. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lee, J.; Kim, K.-J.; Sung, Y.; Park, K.-H.; Oh, E.; Park, C.; Son, Y.-J.; Kang, H. Austalides, osteoclast differentiation inhibitors from a marine-derived strain of the fungus Penicillium rudallense. J. Nat. Prod. 2019, 82, 3083–3088. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Xu, W.; Wang, Y.; Bai, S.; Liu, L.; Luo, Z.; Yuan, W.; Li, Q. Two new meroterpenoids and two new monoterpenoids from the deep sea-derived fungus Penicillium sp. YPGA11. Fitoterapia 2019, 133, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Li, Y.; Xu, W.; Liu, W.; Liu, L.; Zhu, D.; Kang, Y.; Luo, Z.; Li, Q. Three new cyclopiane-type diterpenes from a deep-sea derived fungus Penicillium sp. YPGA11 and their effects against human esophageal carcinoma cells. Bioorganic Chem. 2019, 91, 103129. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Li, Y.; Liu, W.; Liu, L.; Liu, J.; Yuan, W.; Luo, Z.; Xu, W.; Li, Q. Butenolide Derivatives with α-glucosidase inhibitions from the deep-sea-derived fungus Aspergillus terreus YPGA10. Mar. Drugs 2019, 17, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Xu, W.; Liu, L.; Li, S.; Yuan, W.; Luo, Z.; Zhang, J.; Cheng, Y.; Li, Q. Peniginsengins B–E, New farnesylcyclohexenones from the deep sea-derived fungus Penicillium sp. YPGA11. Mar. Drugs 2018, 16, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jim-Min, F.; Yi-Chien, S.; Yu-Hung, C.; Yu-Shia, C. Diterpenes from the bark of Juniperus chinensis. Phytochemistry 1993, 34, 1581–1584. [Google Scholar] [CrossRef]
- Kiren, Y.; Nugroho, A.E.; Hirasawa, Y.; Shirota, O.; Bekenova, M.; Narbekovich, N.O.; Shapilova, M.; Maeno, H.; Morita, H. Mumic acids A–E: New diterpenoids from mumiyo. J. Nat. Med. 2014, 68, 199–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]

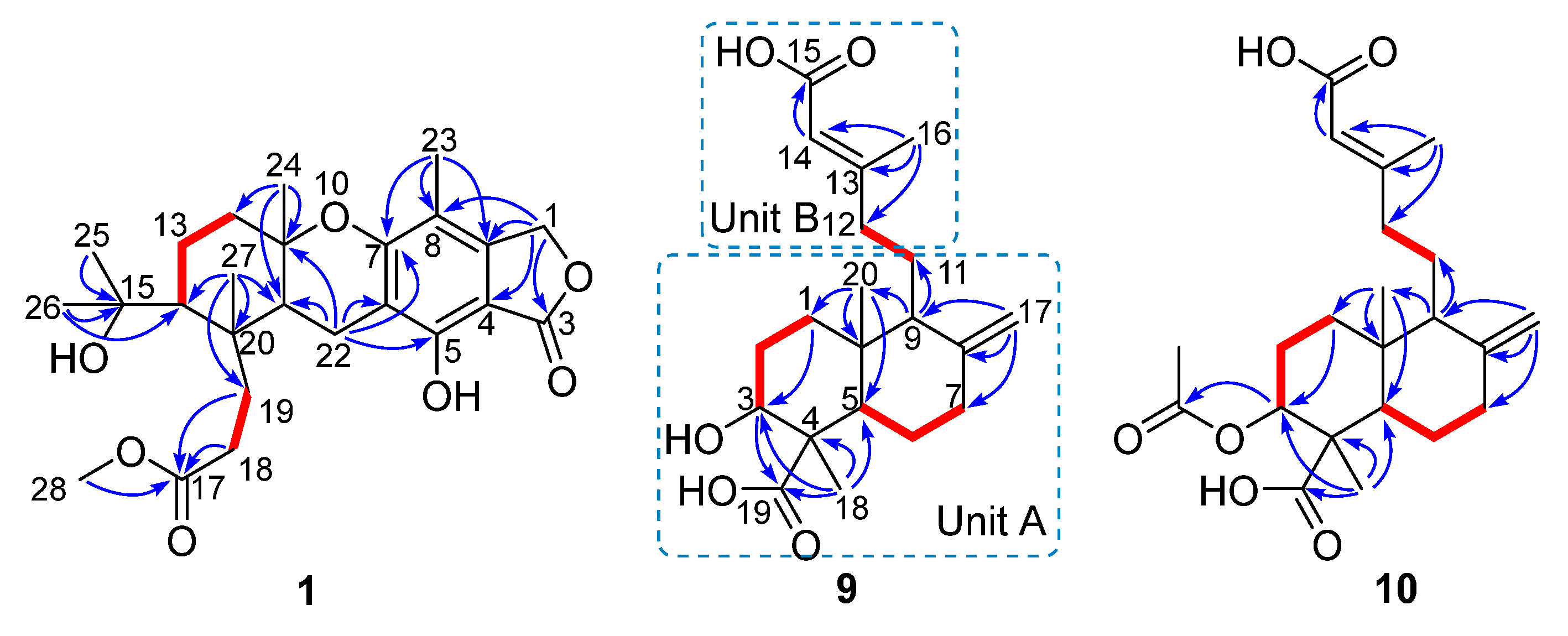
 ) and heteronuclear multiple-bond correlations (HMBC,
) and heteronuclear multiple-bond correlations (HMBC,  ) of compounds 1, 9, and 10.
) and heteronuclear multiple-bond correlations (HMBC, ) of compounds 1, 9, and 10.
) of compounds 1, 9, and 10.
) and heteronuclear multiple-bond correlations (HMBC, ) of compounds 1, 9, and 10.
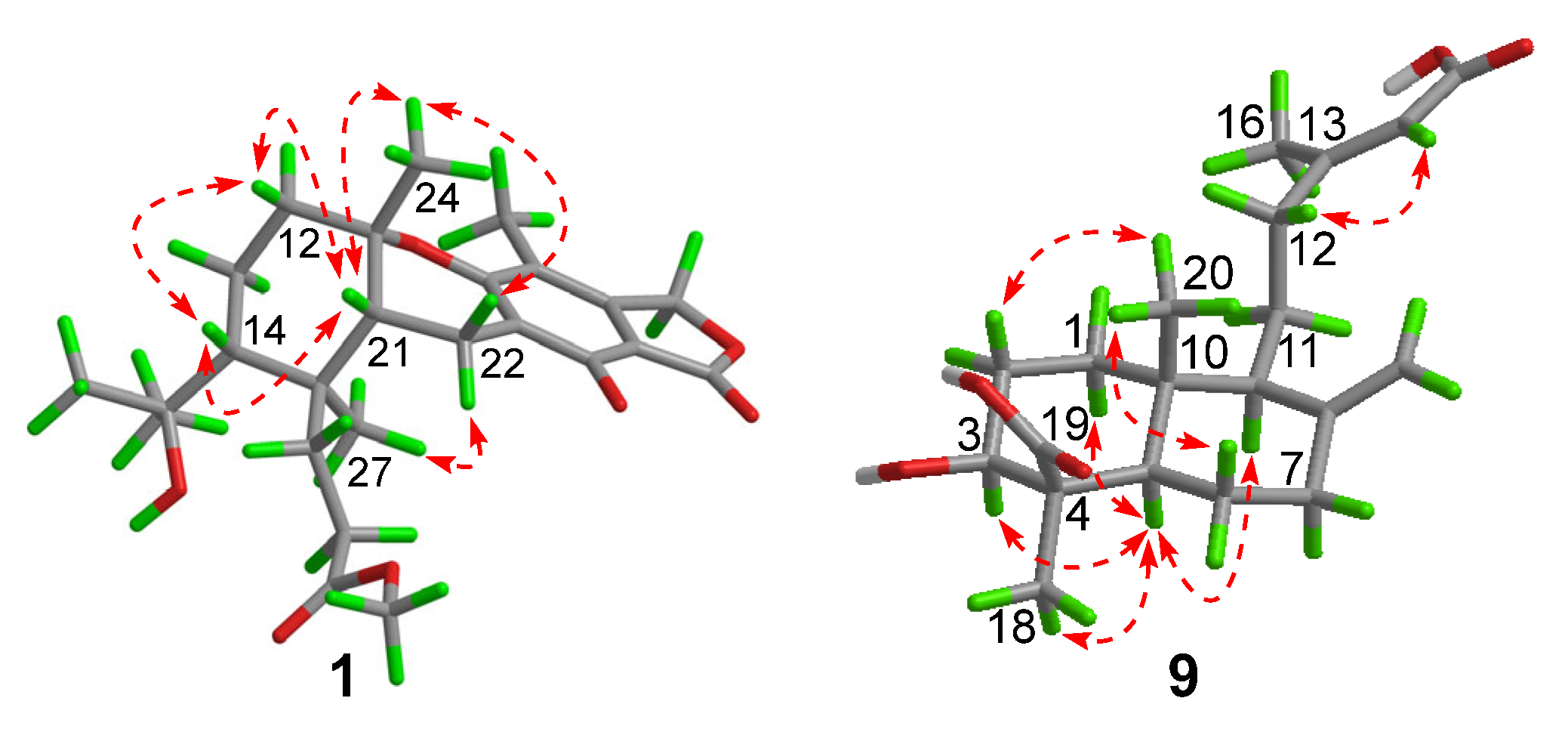
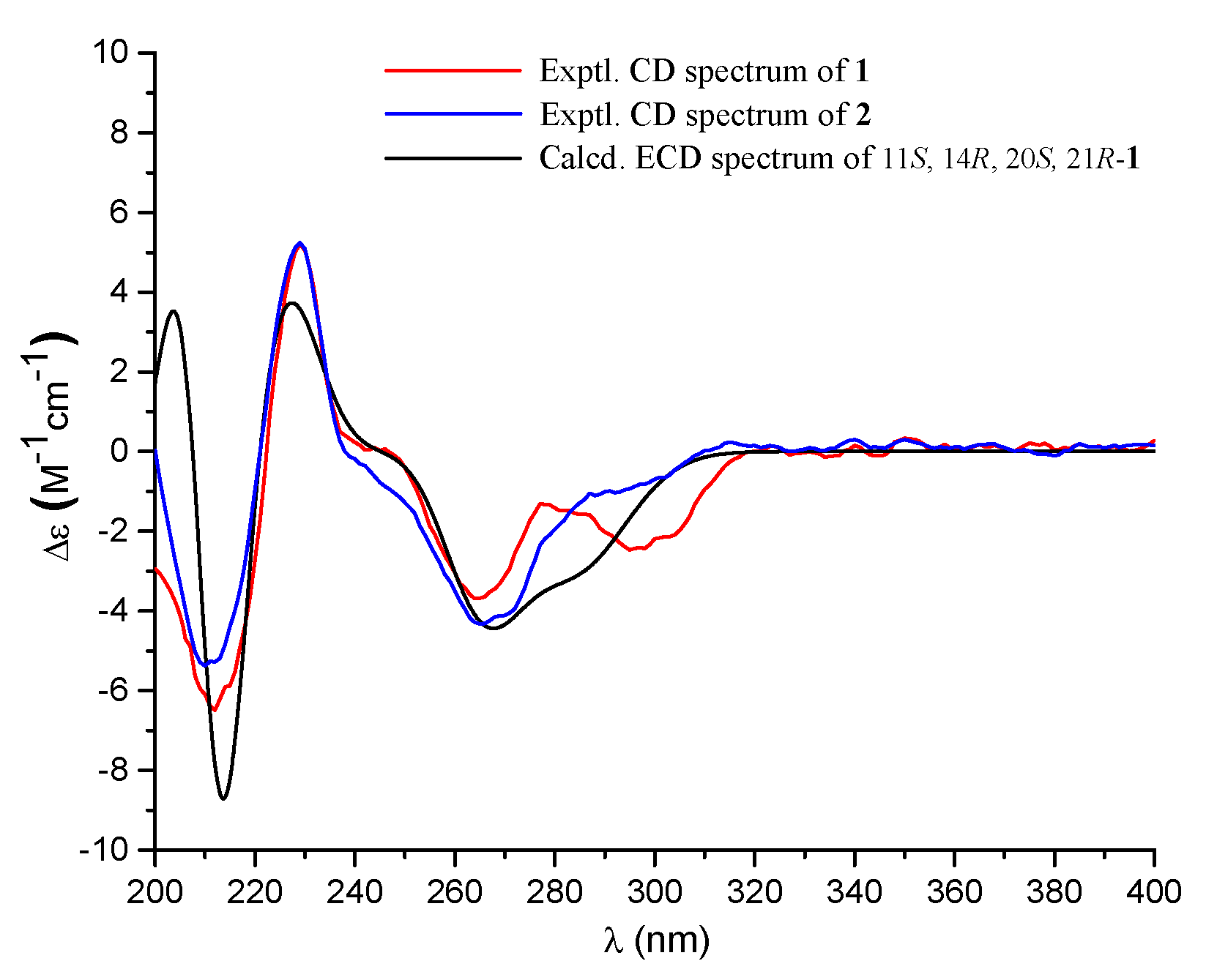
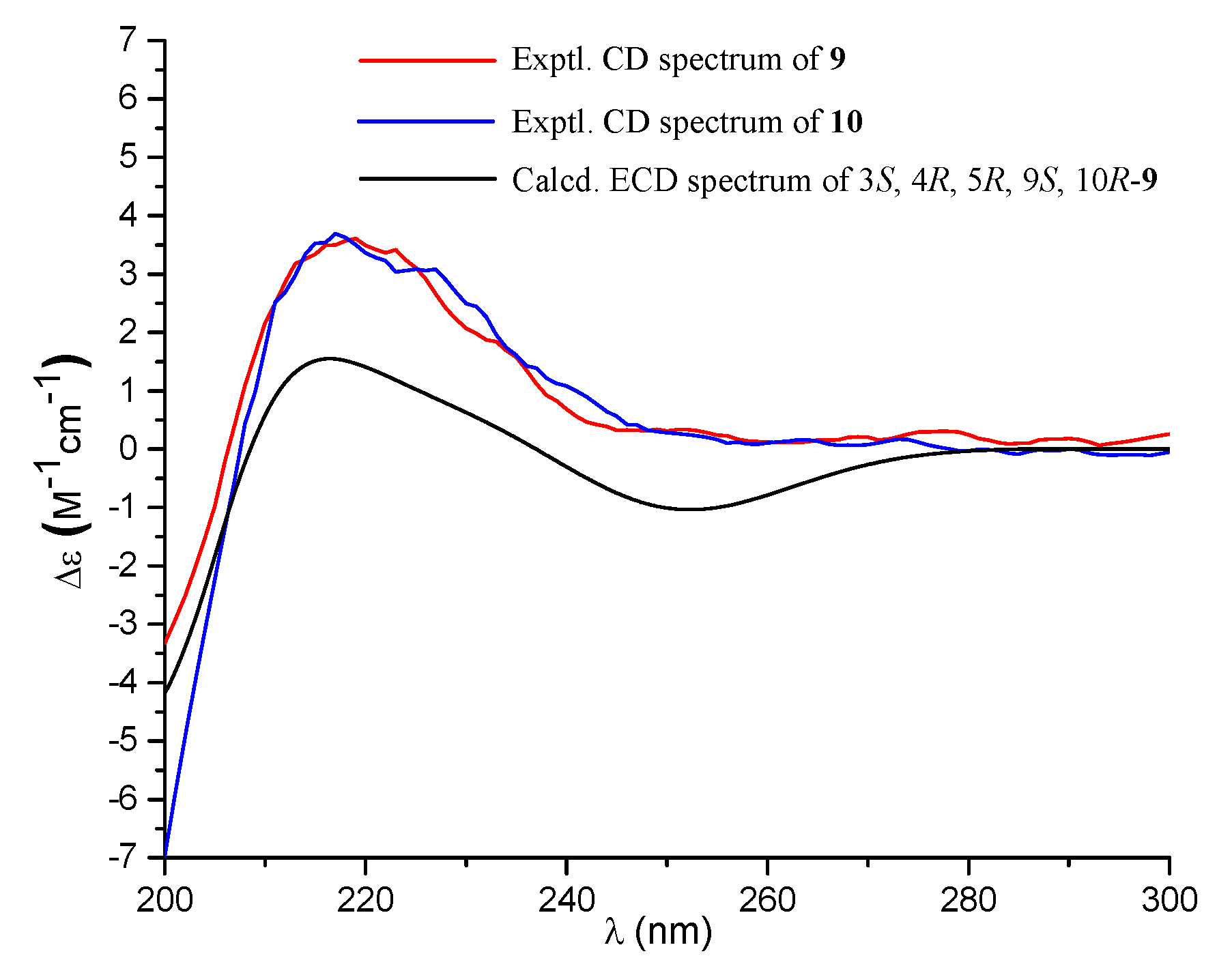
 ) of compounds 1 and 9.
) of compounds 1 and 9.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 3 | 4 | 7 | ||
|---|---|---|---|---|---|
| No. | δH (mult., J in Hz) | δC | δC | δC | δC |
| 1, CH2 | 5.22, s | 70.6 | 69.8 | 69.8 | 69.8 |
| 3, C | 174.2 | 171.9 | 171.9 | 171.8 | |
| 4, C | 103.0 | 108.5 | 108.2 | 108.7 | |
| 5, C | 160.8 | 156.6 | 156.6 | 156.7 | |
| 6, C | 111.2 | 117.5 | 117.4 | 117.3 | |
| 7, C | 153.8 | 159.7 | 160.5 | 159.5 | |
| 8, C | 112.3 | 116.6 | 115.8 | 116.1 | |
| 9, C | 145.4 | 147.5 | 147.5 | 147.0 | |
| 11, C | 78.2 | 78.4 | 78.3 | 79.2 | |
| 12, CH2 | 2.12, m 1.63, m | 40.5 | 45.9 | 40.5 | 45.9 |
| 13, CH2/CH | 1.84, m 1.52, m | 22.6 | 70.1 | 22.6 | 72.2 |
| 14, CH | 1.55, m | 52.1 | 51.7 | 51.9 | 53.4 |
| 15, C | 75.8 | 78.4 | 75.8 | 147.5, | |
| 17, C | 177.0 | 178.6 | 178.9 | 177.9 | |
| 18, CH2 | 2.32, m 2.61, m | 30.1 | 30.5 | 30.3 | 30.0 |
| 19, CH2 | 2.40, m 1.82, m | 34.9 | 36.2 | 35.0 | 36.2 |
| 20, C | 42.8 | 42.1 | 42.8 | 40.7 | |
| 21, CH | 1.68, d (8.2) | 41.5 | 41.5 | 41.4 | 40.7 |
| 22, CH2 | 2.69, dd (18.3, 8.2) 2.94, d (18.3) | 18.4 | 18.7 | 18.8 | 18.9 |
| 23, CH3 | 2.02, s | 10.6 | 10.8 | 10.6 | 10.8 |
| 24, CH3 | 1.21, s | 27.7 | 28.2 | 27.8 | 27.9 |
| 25, CH3/CH2 | 1.28, s | 33.2 | 32.1 | 33.2 | 116.1 |
| 26, CH3 | 1.21, s | 28.2 | 32.7 | 28.2 | 26.5 |
| 27, CH3 | 0.71, s | 19.4 | 18.5 | 19.6 | 20.9 |
| COOCH3 | 3.67, s | 52.0 | 52.1 | ||
| 5-OCH3 | 62.3 | 62.2 | 62.3 | ||
| No. | 9 | 10 | ||
|---|---|---|---|---|
| δH (mult., J in Hz) | δC | δH (mult., J in Hz) | δC | |
| 1, CH2 | 1.24, m 1.91, m | 38.7 | 1.31, m 1.93, m | 38.1 |
| 2, CH2 | 1.78, m 2.16, m | 29.7 | 1.72, m 2.46, m | 25.7 |
| 3, CH | 3.18, dd (12.1, 4.4) | 79.0 | 4.55, dd (12.3, 4.5) | 80.9 |
| 4, C | 50.3 | 49.3 | ||
| 5, CH | 1.29, m | 56.5 | 1.43, dd (12.8, 2.4) | 56.6 |
| 6, CH2 | 1.93, m 2.03, m | 27.2 | 1.63, m 2.04, m | 26.9 |
| 7, CH2 | 1.93, m 2.42, m | 39.6 | 1.99, m 2.43, m | 39.3 |
| 8, C | 149.0 | 148.7 | ||
| 9, CH | 1.61, m | 56.5 | 1.65, m | 56.4 |
| 10, C | 41.0 | 40.8 | ||
| 11, CH2 | 1.73, m 1.58, m | 23.0 | 1.75, m 1.58, m | 23.0 |
| 12, CH2 | 2.02, m 2.30, m | 40.7 | 2.03, m 2.30, m | 40.6 |
| 13, C | 161.7 | 161.2 | ||
| 14, CH | 5.61, s | 116.9 | 5.62, s | 115.8 |
| 15, C | 170.4 | 170.7 | ||
| 16, CH3 | 2.13, d (1.0) | 18.9 | 2.13, s | 18.9 |
| 17, CH2 | 4.56, br s 4.90, br s | 107.3 | 4.57, br s 4.92, br s | 107.6 |
| 18, CH3 | 1.38, s | 24.8 | 1.24, s | 24.7 |
| 19, C | 180.5 | 177.5 | ||
| 20, CH3 | 0.69, s | 13.5 | 0.74, s | 13.1 |
| COCH3 | 2.04, s | 21.1 | ||
| COCH3 | 172.8 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Z.; Liu, W.; Fan, R.; Han, S.; Li, Y.; Cui, X.; Zhang, J.; Wu, Y.; Lv, X.; Zhang, Y.; et al. Terpenoids from the Deep-Sea-Derived Fungus Penicillium thomii YPGA3 and Their Bioactivities. Mar. Drugs 2020, 18, 164. https://doi.org/10.3390/md18030164
Cheng Z, Liu W, Fan R, Han S, Li Y, Cui X, Zhang J, Wu Y, Lv X, Zhang Y, et al. Terpenoids from the Deep-Sea-Derived Fungus Penicillium thomii YPGA3 and Their Bioactivities. Marine Drugs. 2020; 18(3):164. https://doi.org/10.3390/md18030164
Chicago/Turabian StyleCheng, Zhongbin, Wan Liu, Runzhu Fan, Shouye Han, Yuanli Li, Xiaoyun Cui, Jia Zhang, Yinan Wu, Xin Lv, Yun Zhang, and et al. 2020. "Terpenoids from the Deep-Sea-Derived Fungus Penicillium thomii YPGA3 and Their Bioactivities" Marine Drugs 18, no. 3: 164. https://doi.org/10.3390/md18030164