Polyketide Derivatives from Mangrove Derived Endophytic Fungus Pseudopestalotiopsis theae

 , , and
, , and
Abstract
:1. Introduction
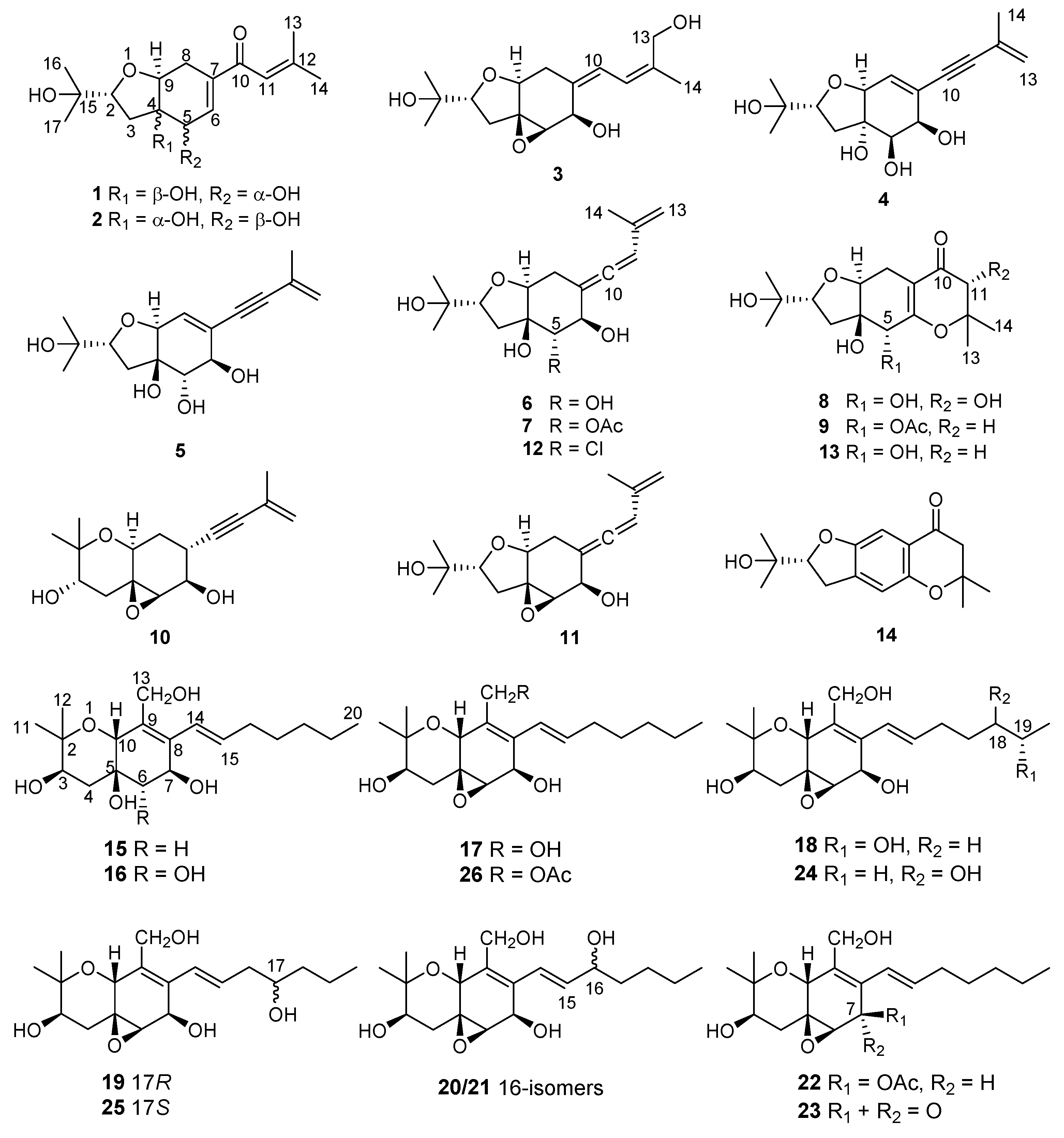
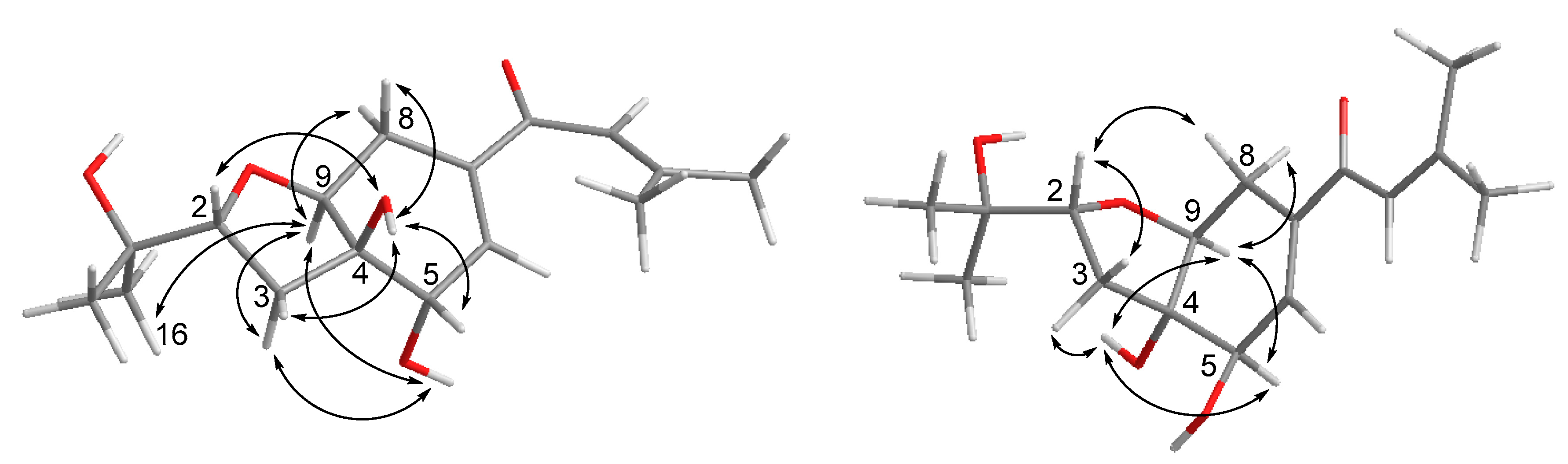
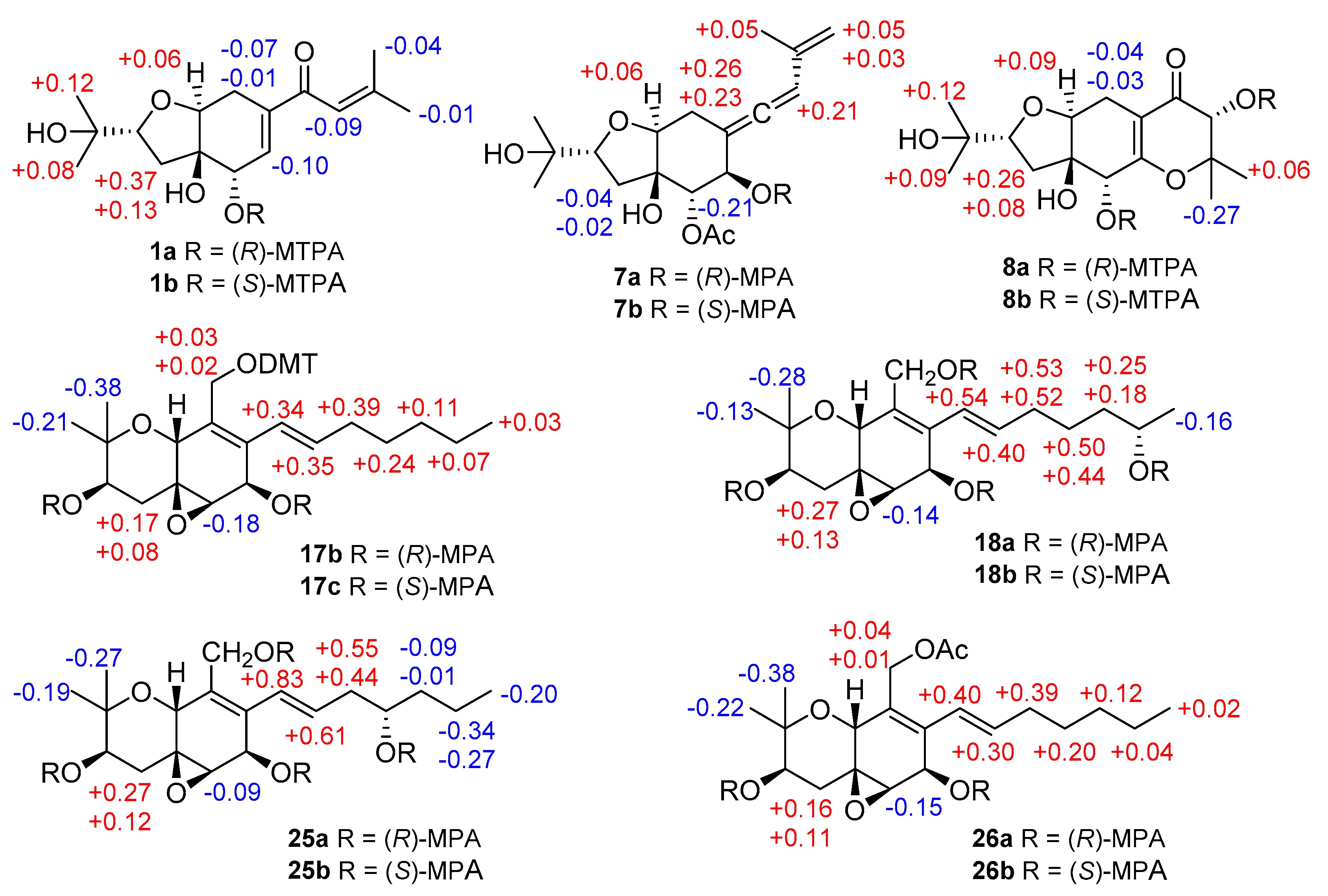
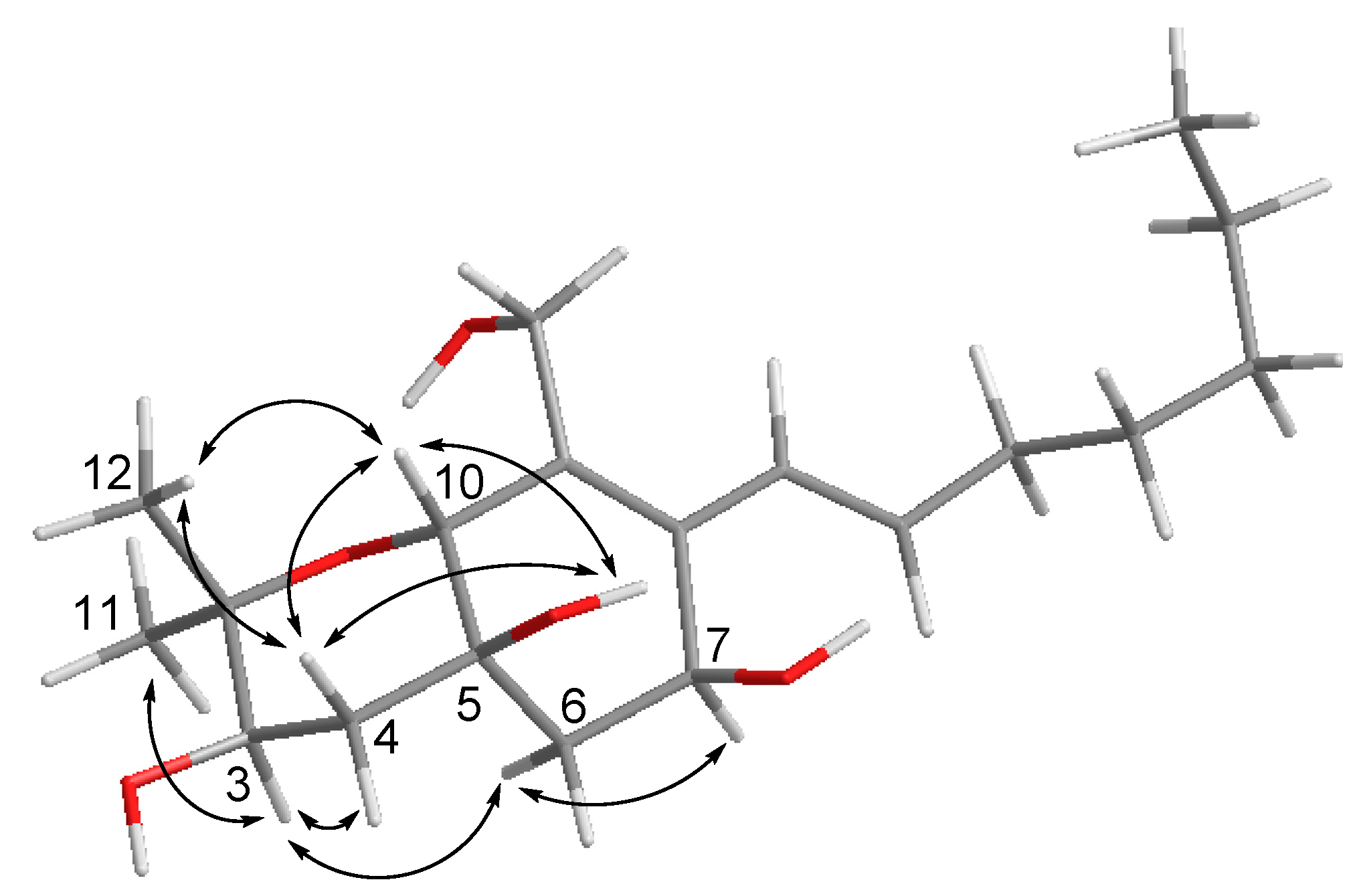
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation, Extraction, and Isolation
3.4. Preparation of (R)- and (S)-MTPA Esters
3.5. Preparation of Compound 17a
3.6. Preparation of (R)- and (S)-MPA Esters
3.7. Cytotoxicity and Antibacterial Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Surup, F.; Stadler, M. Natural products in the chemical industry. Von Bernd Schaefer. Angew. Chem. 2015, 127, 8999–9000. [Google Scholar] [CrossRef]
- Petersen, A.B.; Rønnest, M.H.; Larsen, T.O.; Clausen, M.H. The Chemistry of Griseofulvin. Chem. Rev. 2014, 114, 12088–12107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobert, J.A. Lovastatin and beyond: The history of the HMG-CoA reductase inhibitors. Nat. Rev. Drug Discov. 2003, 2, 517–526. [Google Scholar] [CrossRef]
- Watts, R.; Clunie, G.; Hall, F.; Marshall, T. Rheumatology; Oxford University Press: New York, NY, USA, 2009; p. 558. [Google Scholar]
- Kusari, S.; Pandey, S.P.; Spiteller, M. Untapped mutualistic paradigms linking host plant and endophytic fungal production of similar bioactive secondary metabolites. Phytochem. 2013, 91, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, S.; Prakash, V.; Ranjan, N. Recent advances in the discovery of bioactive metabolites from Pestalotiopsis. Phytochem. Rev. 2017, 71, 882. [Google Scholar] [CrossRef]
- Maharachchikumbura, S.; Hyde, K.; Groenewald, J.; Xu, J.; Crous, P. Pestalotiopsis revisited. Stud. Mycol. 2014, 79, 121–186. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Ebada, S.S.; Proksch, P. Pestalotiopsis a highly creative genus: Chemistry and bioactivity of secondary metabolites. Fungal Divers. 2010, 44, 15–31. [Google Scholar] [CrossRef]
- Yang, X.-L.; Zhang, J.-Z.; Luo, D.-Q. The taxonomy, biology and chemistry of the fungal Pestalotiopsis genus. Nat. Prod. Rep. 2012, 29, 622. [Google Scholar] [CrossRef]
- Li, E.; Jiang, L.; Guo, L.; Zhang, H.; Che, Y. Pestalachlorides A–C, antifungal metabolites from the plant endophytic fungus Pestalotiopsis adusta. Bioorganic Med. Chem. 2008, 16, 7894–7899. [Google Scholar] [CrossRef]
- Liu, L.; Liu, S.; Jiang, L.; Chen, X.; Guo, L.; Che, Y. ChemInform Abstract: Chloropupukeananin, the First Chlorinated Pupukeanane Derivative, and Its Precursors from Pestalotiopsis fici. Chemin 2008, 39, 1397–1400. [Google Scholar] [CrossRef]
- Ding, G.; Zhang, F.; Chen, H.; Guo, L.; Zou, Z.; Che, Y. Pestaloquinols A and B, Isoprenylated Epoxyquinols fromPestalotiopsissp. J. Nat. Prod. 2011, 74, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Ge, H.M.; Li, F.; Song, Y.C.; Tan, R.X. New Phytotoxic Metabolites fromPestalotiopsissp. HC02, a Fungus Residing inChondracris roseeGut. Chem. Biodivers. 2008, 5, 2402–2407. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Dai, H.; Makhloufi, G.; Heering, C.; Janiak, C.; Hartmann, R.; Mandi, A.; Kurtán, T.; Müller, W.E.G.; Kassack, M.U.; et al. Cytotoxic 14-Membered Macrolides from a Mangrove-Derived Endophytic Fungus, Pestalotiopsis microspora. J. Nat. Prod. 2016, 79, 2332–2340. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, N.; Proksch, P.; Zhou, D.; Lin, W. Truncateols O-V, further isoprenylated cyclohexanols from the sponge-associated fungus Truncatella angustata with antiviral activities. Phytochemistry 2018, 155, 61–68. [Google Scholar] [CrossRef]
- Zhao, Y.; Si, L.; Liu, N.; Proksch, P.; Zhou, D.; Lin, W. Truncateols A–N, new isoprenylated cyclohexanols from the sponge-associated fungus Truncatella angustata with anti-H1N1 virus activities. Tetrahedron 2015, 71, 2708–2718. [Google Scholar] [CrossRef]
- Li, E.; Tian, R.; Liu, S.; Chen, X.; Guo, L.; Che, Y. Pestalotheols A–D, Bioactive Metabolites from the Plant Endophytic FungusPestalotiopsis theae. J. Nat. Prod. 2008, 71, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Ciavatta, M.L.; López-Gresa, M.P.; Gavagnin, M.; Nicoletti, R.; Manzo, E.; Mollo, E.; Guo, Y.-W.; Cimino, G. Cytosporin-related compounds from the marine-derived fungus Eutypella scoparia. Tetrahedron 2008, 64, 5365–5369. [Google Scholar] [CrossRef]
- Akone, S.H.; El Amrani, M.; Lin, W.; Lai, D.; Proksch, P. Cytosporins F–K, new epoxyquinols from the endophytic fungus Pestalotiopsis theae. Tetrahedron Lett. 2013, 54, 6751–6754. [Google Scholar] [CrossRef]
- Stevens-Miles, S.; Goetz, M.A.; Bills, G.; Giacobbe, R.A.; Tkacz, J.S.; Chang, R.S.L.; Mojena, M.; Martín, I.; Díez, M.T.; Pelae, F.; et al. Discovery of an Angiotensin II Binding Inhibitor from a Cytospora sp. Using Semi-automated Screening Procedures. J. Antibiot. 1996, 49, 119–123. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.-X.; Sun, D.-W.; Zheng, C.-J.; Wang, C. A new hexahydrobenzopyran derivative from the gorgonian-derived Fungus Eutypella sp. Nat. Prod. Res. 2017, 31, 1640–1646. [Google Scholar] [CrossRef]
- Rivera-Chávez, J.; Zacatenco-Abarca, J.; Morales-Jiménez, J.; Martínez-Aviña, B.; Hernández-Ortega, S.; Aguilar-Ramírez, E. Cuautepestalorin, a 7,8-Dihydrochromene–Oxoisochromane Adduct Bearing a Hexacyclic Scaffold from Pestalotiopsis sp. IQ-011. Org. Lett. 2019, 21, 3558–3562. [Google Scholar] [CrossRef] [PubMed]
- Kjer, J.; Debbab, A.; Aly, A.H.; Proksch, P. Methods for isolation of marine-derived endophytic fungi and their bioactive secondary products. Nat. Protoc. 2010, 5, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Ashour, M.; Edrada, R.; Ebel, R.; Wray, V.; Wätjen, W.; Padmakumar, K.; Müller, W.E.G.; Lin, W.H.; Proksch, P. Kahalalide Derivatives from the Indian Sacoglossan MolluskElysiagrandifolia. J. Nat. Prod. 2006, 69, 1547–1553. [Google Scholar] [CrossRef] [PubMed]
- Hecht, D.W.; Citron, D.M.; Cox, M.; Jacobus, N.; Jenkins, S.G.; Onderdonk, A.; Wexler, H.M. Methods for Antimicrobial Susceptibiluty Testing of Anaerobic Bacteria: Approved Standard; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2007; pp. 1–47. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 a,c | 3 b | |||
|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 2 | 84.2, CH | 3.95, dd (9.9, 6.4) | 84.1, CH | 3.57, dd (8.3, 5.7) | 86.0, CH | 4.07, dd (9.4, 6.7) |
| 3 | 34.0, CH2 | 2.20, dd (12.4, 9.9) 1.60, dd (12.4, 6.4) | 36.1, CH2 | 2.08, dd (13.5, 8.3) 1.81, dd (13.5, 5.7) | 31.0, CH2 | 2.55, dd, (13.3, 9.4) 1.72, dd, (13.3, 6.7) |
| 4 | 77.8, C | 82.5, C | 69.5, C | |||
| 5 | 68.9, CH | 4.05, dd (5.3, 4.6) | 72.3, CH | 4.24, dd (5.0, 2.5) | 60.1, CH | 3.75, d (2.8) |
| 6 | 138.0, CH | 6.71, dd (4.6, 2.3) | 141.0, CH | 6.69, t (2.5) | 64.6, CH | 4.65, dd (8.8, 2.8) |
| 7 | 138.7, C | 137.8, C | 135.5, C | |||
| 8 | 25.7, CH2 | 2.53, dd (16.0, 5.3) 2.09, ddd (16.0, 10.7, 2.3) | 26.8, CH2 | 2.54, dd (16.9, 3.9) 2.14, ddd (16.9, 5.4, 2.5) | 35.4, CH2 | 2.50, dd (12.1, 11.5) 2.21, dd, (12.1, 4.8) |
| 9 | 76.3, CH | 3.63, dd (10.7, 5.3) | 82.7, CH | 4.05, dd (5.4, 3.9) | 77.7, CH | 3.86, dd (11.5, 4.8) |
| 10 | 191.9, C | 190.1, C | 127.6, CH | 6.25, d (11.7) | ||
| 11 | 120.7, CH | 6.57, s | 120.0, CH | 6.59, s | 122.8, CH | 6.29, d (11.7) |
| 12 | 153.7, C | 153.6, C | 139.9, C | |||
| 13 | 20.5, CH3 | 2.00, s | 20.5, CH3 | 2.00, s | 60.9, CH2 | 4.21, dd (12.5, 5.3) 4.16, dd (12.5, 5.3) |
| 14 | 27.2, CH3 | 1.90, s | 27.1, CH3 | 1.90, s | 22.0, CH3 | 1.85, s |
| 15 | 70.8, C | 70.9, C | 71.9, C | |||
| 16 | 25.8, CH3 | 1.07, s | 26.0, CH3 | 1.09, s | 26.6, CH3 | 1.23, s |
| 17 | 25.9, CH3 | 1.01, s | 26.7, CH3 | 0.99, s | 25.8, CH3 | 1.09, s |
| 4-OH | 4.54, s | 5.37, s | ||||
| 5-OH | 5.38, d (5.3) | 5.52, d (5.0) | ||||
| 6-OH | 3.97, d (8.8) | |||||
| 13-OH | 3.70, t (5.3) | |||||
| 15-OH | 4.16, s | 4.75, s | 3.34, s | |||
| No. | 4 a | 5 a,b | 6 a | |||
|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 2 | 85.2, CH | 3.73, dd (8.1, 7.7) | 84.6, CH | 4.01, dd (9.6, 6.4) | 83.7, CH | 3.86, dd (9.9, 6.3) |
| 3 | 37.5, CH2 | 2.43, dd (13.0, 7.7) 1.79, dd (13.0, 8.1) | 33.5, CH2 | 2.12, (dd, 12.0, 9.6) 1.59, dd, (12.0, 6.4) | 34.1, CH2 | 2.12, dd (12.0, 9.9) 1.53, dd (12.0, 6.3) |
| 4 | 79.1, C | 76.4, C | 79.8, C | |||
| 5 | 71.8, CH | 3.60, t (4.0) | 72.5, CH | 3.81, d (3.7) | 71.1, CH | 3.83, d (3.8) |
| 6 | 67.6, CH | 4.11, dd (9.0, 4.0) | 74.1, CH | 3.73, d (9.1) | 74.9, CH | 4.01, s |
| 7 | 124.1, C | 122.2, C | 102.2, C | |||
| 8 | 134.0, CH | 5.87, d (3.2) | 132.9, CH | 6.10, d (1.7) | 27.8, CH2 | 2.52, ddd (12.4, 11.9, 3.9) 2.23, dd (11.9, 4.4) |
| 9 | 81.2, CH | 4.10, d (3.2) | 77.1, CH | 4.35, d (1.7) | 77.4, CH | 3.81, dd (12.4, 4.4) |
| 10 | 88.6, C | 88.8, C | 204.6, C | |||
| 11 | 90.2, C | 88.7, C | 95.7, CH | 5.92, d (3.9) | ||
| 12 | 126.4, C | 126.0, C | 139.0, C | |||
| 13 | 122.1, CH2 | 5.32, s 5.27, s | 121.6, CH2 | 5.30, s 5.26, s | 113.5, CH2 | 4.93, s 4.82, s |
| 14 | 23.2, CH3 | 1.87, s | 23.0, CH3 | 1.87, s | 19.5, CH3 | 1.69, s |
| 15 | 70.2, C | 70.5, C | 70.5, C | |||
| 16 | 25.9, CH3 | 1.07, s | 25.8, CH3 | 1.10, s | 26.1, CH3 | 1.07, s |
| 17 | 26.4, CH3 | 1.00, s | 25.5, CH3 | 0.99, s | 25.9, CH3 | 0.98, s |
| 4-OH | 5.05, s | 4.68, s | 5.22, s | |||
| 5-OH | 4.77, d (4.0) | 5.47, d (3.7) | 5.28, d (3.8) | |||
| 6-OH | 5.08, d (9.0) | 4.72, d (9.1) | 5.74, br s | |||
| 15-OH | 4.31, s | 4.23, s | 4.17, s | |||
| No. | 7 a | 8 b | 9 a,c | |||
|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 2 | 83.5, CH | 3.88, dd (9.6, 6.4) | 86.1, CH | 4.13, dd (10.1, 6.4) | 84.4, CH | 3.95, dd (9.5, 6.5) |
| 3 | 34.0, CH | 1.83, dd (12.3, 9.6) 1.62, dd (12.3, 6.4) | 35.0, CH2 | 2.49, dd (12.5, 10.1) 1.77, dd (12.5, 6.4) | 33.9, CH2 | 1.80, dd (12.5, 9.5) 1.66, dd (12.5, 6.5) |
| 4 | 77.8, C | 80.1, C | 76.4, C | |||
| 5 | 72.0, CH | 5.01, d (2.5) | 72.2, CH | 4.08, d (5.1) | 71.3, CH | 5.28, s |
| 6 | 71.7, CH | 4.04, d (2.5) | 168.3, C | 162.3, C | ||
| 7 | 101.5, C | 107.0, C | 110.0, C | |||
| 8 | 27.4, CH2 | 2.56, ddd (12.4, 12.0, 3.9,) 2.31, dd (12.0, 4.4) | 22.4, CH2 | 2.70, dd (14.9, 5.7) 2.11 (dd, 14.9, 10.6) | 21.3, CH2 | 2.55, dd (14.9, 5.5) 2.01, dd (14.9, 10.7) |
| 9 | 78.0, CH | 3.76, dd (12.4. 4.4) | 76.9, CH | 3.91, dd (10.6, 5.7) | 76.5, CH | 3.63, dd (10.7, 5.5) |
| 10 | 204.4, CH | 194.6, C | 191.9, C | |||
| 11 | 96.5, CH | 6.02, d (3.9) | 76.1, CH | 4.13, d (2.8) | 46.4, CH2 | 2.65, d (16.7) 2.42, d (16.7) |
| 12 | 138.2, C | 84.4, C | 80.4, C | |||
| 13 | 114.5, CH2 | 4.99, s 4.88, s | 26.9, CH3 | 1.49, s | 27.3, CH3 | 1.36, s |
| 14 | 19.5, CH3 | 1.71, s | 16.5, CH3 | 1.14, s | 23.4, CH3 | 1.24, s |
| 15 | 70.5, C | 72.0, C | 70.5, C | |||
| 16 | 25.9, CH3 | 1.09, s | 26.7, CH3 | 1.18, s | 26.1, CH3 | 0.99, s |
| 17 | 26.0, CH3 | 0.98, s | 25.6, CH3 | 1.09, s | 25.6, CH3 | 1.08, s |
| 4-OH | 5.45, s | 3.97, s | 5.31, s | |||
| 5-OH | 5.09, d (5.1) | |||||
| 6-OH | 5.91, br s | |||||
| 11-OH | 4.30, d (2.8) | |||||
| 13-OH | 4.24, s | |||||
| 15-OH | 3.22, s | 4.25, s | ||||
| 5-OAc | 20.6, CH3 169.2, C | 2.00, s | 20.5, CH3 169.5, C | 2.09, s | ||
| No. | 15 a | 16 a | 17 b | |||
|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 2 | 74.9, C | 75.0, C | 75.4, C | |||
| 3 | 70.6, CH | 3.20, dt (12.0, 4.9) | 69.8, CH | 3.78, dt (11.4, 5.5) | 72.1, CH | 3.38, dt (11.7, 5.0) |
| 4 | 42.7, CH2 | 1.75, dd (12.8, 4.9) 1.65, dd (12.8, 12.0) | 42.3, CH2 | 2.00, dd (13.2, 5.5) 1.68, dd (13.2, 11.4) | 35.8, CH2 | 2.04, dd (12.7, 11.7) 1.50, dd (12.7, 5.0) |
| 5 | 69.3, C | 69.2, C | 58.1, C | |||
| 6 | 35.7, CH2 | 2.00, dd (13.9, 4.3) 1.54, d (13.9) | 75.3, CH | 3.55, d (8.0) | 61.8, CH | 3.24, d (2.7) |
| 7 | 63.9, CH | 4.30, dd (7.6, 4.3) | 69.5, CH | 4.11, d (7.6) | 64.8, CH | 4.47, dd (7.3, 2.7) |
| 8 | 133.8, C | 131.8, C | 132.9, C | |||
| 9 | 132.0, C | 131.7, C | 130.0, C | |||
| 10 | 70.0, CH | 3.87, s | 70.1, CH | 4.01, s | 65.4, CH | 4.34, s |
| 11 | 27.9, CH3 | 1.07, s | 27.7, CH3 | 1.06, s | 27.7, CH3 | 1.12, s |
| 12 | 16.1, CH3 | 1.10, s | 16.4, CH3 | 1.13, s | 16.1, CH3 | 1.16, s |
| 13 | 57.7, CH2 | 4.21, dd (11.9, 4.0) 3.81, dd (11.9, 6.2) | 57.5, CH2 | 4.24, dd (11.9, 4.2) 3.92, dd (11.9, 6.2) | 58.0, CH2 | 4.09, br d (11.6) 3.79, br d (11.6) |
| 14 | 126.0, CH | 6.36, d (15.7) | 126.4, CH | 6.44, d (15.8) | 124.9, CH | 6.06, d (15.8) |
| 15 | 132.1, CH | 5.90, dt (15.7, 7.0) | 132.1, CH | 5.91, dt (15.8, 7.0) | 134.5, CH | 5.85, dt (15.8, 6.8) |
| 16 | 32.8, CH2 | 2.10, m | 32.8, CH2 | 2.12, m | 33.0, CH2 | 2.06, m |
| 17 | 28.6, CH2 | 1.37, m | 28.6, CH2 | 1.38, m | 28.5, CH2 | 1.36, m |
| 18 | 30.8, CH2 | 1.27, m | 30.8, CH2 | 1.27, m | 30.8, CH2 | 1.27, m |
| 19 | 22.0, CH2 | 1.29, m | 22.0, CH2 | 1.29, m | 22.0, CH2 | 1.28, m |
| 20 | 13.9, CH3 | 0.86, t (6.9) | 13.9, CH3 | 0.87, t (6.9) | 13.9, CH3 | 0.86, t (7.0) |
| 3-OH | 4.76, d (4.9) | 4.70, d (5.5) | 5.05, d (5.0) | |||
| 5-OH | 5.07, s | 5.16, s | ||||
| 6-OH | 3.98, d (8.0) | |||||
| 7-OH | 4.75, d (7.6) | 4.88, d (7.6) | 4.92, d (7.3) | |||
| 13-OH | 4.53, dd (6.2, 4.0) | 4.54, dd (6.2, 4.2) | 4.55, br s | |||
| No. | 18 a | 19 b,c | 20 b,c | |||
|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 2 | 75.4, C | 75.2, C | 75.2, C | |||
| 3 | 72.1, CH | 3.39, m | 71.8, CH | 3.38, dt (11.8, 5.0) | 71.8, CH | 3.38, dt (11.8, 4.9) |
| 4 | 35.8, CH2 | 2.04, m 1.50, dd (12.7, 5.0) | 35.5, CH2 | 2.05, dd (12.6, 11.8) 1.50, dd (12.6, 5.0) | 35.5, CH2 | 2.05, dd (12.7, 11.8) 1.50, dd (12.7, 4.9) |
| 5 | 58.1, C | 57.7, C | 57.7, C | |||
| 6 | 61.8, CH | 3.24, d (2.5) | 61.5, CH | 3.25, d (2.6) | 61.5, CH | 3.25, d (2.5) |
| 7 | 64,8, CH | 4.48, dd (7.6, 2.5) | 64.5, CH | 4.48, dd (7.4, 2.6) | 64.4, CH | 4.49, d (7.5, 2.5) |
| 8 | 132.9, C | 132.5, C | 132.3, C | |||
| 9 | 130.0, C | 129.8, C | 130.4, C | |||
| 10 | 65.4, CH | 4.34, s | 65.1, CH | 4.34, s | 65.0, CH | 4.35, s |
| 11 | 27.7, CH3 | 1.12, s | 27.5, CH3 | 1.12, s | 27.4, CH3 | 1.12, s |
| 12 | 16.1, CH3 | 1.16, s | 15.8, CH3 | 1.16, s | 15.8, CH3 | 1.16, s |
| 13 | 58.0, CH2 | 4.09, dd (12.0, 2.0) 3.79, dd (12.0, 3.5) | 57.7, CH2 | 4.11, dd (11.9, 2.3) 3.79, dd (11.9, 4.2) | 57.6, CH2 | 4.12, dd (11.9, 2.0) 3.79, dd (11.9, 4.0) |
| 14 | 124.9, CH | 6.06, d (15.9) | 126.2, CH | 6.08, d (16.0) | 122.8, CH | 6.20, d (16.0) |
| 15 | 134.6, CH | 5.85, dt (15.9, 7.0) | 131.4, CH | 5.88, dt (16.0, 7.2) | 137.8, CH | 5.86, dd (16.0, 6.2) |
| 16 | 33.1, CH2 | 2.05, m | 41.5, CH2 | 2.15, m | 71.0, CH2 | 3.95, m |
| 17 | 25.2, CH2 | 1.40, m 1.34, m | 69.2, CH | 3.46, m | 36.9, CH2 | 1.40, m |
| 18 | 38.5, CH2 | 1.33, m 1.30, m | 38.4, CH2 | 1.36, m 1.27, m | 27.0, CH2 | 1.28, m |
| 19 | 65.6, CH | 3.57, m | 18.1, CH2 | 1.38, m 1.27, m | 21.9, CH2 | 1.27, m |
| 20 | 23.7, CH3 | 1.03, d (6.1) | 13.8, CH3 | 0.85, t (6.9) | 13.7, CH3 | 0.86, t (6.9) |
| 3-OH | 5.06, d (4.5) | 5.07, d (5.0) | 5.07, d (4.9) | |||
| 7-OH | 4.94, d (7.6) | 4.92, d (7.4) | 4.94, d (7.5) | |||
| 13-OH | 4.56, dd (3.5, 2.0) | 4.56, dd (4.2, 2.3) | 4.58, dd (4.0, 2.0) | |||
| 16-OH | 4.62, d (4.0) | |||||
| 17-OH | 4.34, br s | |||||
| 19-OH | 4.31, d (4.3) | |||||
| No. | 21 a | 22 b | 23 b,c | |||
|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 2 | 75.4, C | 75.7, C | 76.7, C | |||
| 3 | 72.0, CH | 3.39, dt (11.8, 4.9) | 71.8, CH | 3.42, dt (11.8, 4.9) | 71.9, CH | 3.45, dt (11.6, 5.0) |
| 4 | 35.8, CH2 | 2.05, dd (12.6, 11.8) 1.50, dd (12.6, 4.9) | 35.2, CH2 | 2.06, dd (12.7, 11.8) 1.53, dd (12.7, 4.9) | 34.0, CH2 | 2.21, dd (12.8, 11.6) 1.63, dd (12.8, 5.0) |
| 5 | 57.9, C | 58.0, C | 61.1, C | |||
| 6 | 61.7, CH | 3.25, d (2.3) | 58.1, CH | 3.39, d (2.8) | 59.3, CH | 3.54, d (1.2) |
| 7 | 64.8, CH | 4.48, dd (7.3, 2.3) | 67.6, CH | 5.85, d (2.8) | 195.8, C | |
| 8 | 132.5, C | 128.0, C | 130.3, C | |||
| 9 | 130.7, C | 133.4, C | 148.2, C | |||
| 10 | 65.3, CH | 4.35, s | 64.7, CH | 4.40, s | 64.5, CH | 4.74, d (1.2) |
| 11 | 27.7, CH3 | 1.12, s | 27.6, CH3 | 1.13, s | 27.5, CH3 | 1.16, s |
| 12 | 16.1, CH3 | 1.16, s | 16.1, CH3 | 1.17, s | 16.1, CH3 | 1.24, s |
| 13 | 58.0, CH2 | 4.12, dd (11.8, 4.1) 3.79, dd (11.8, 5.0) | 57.6, CH2 | 4.11, dd (12.0, 4.4) 3.78, dd (12.0, 5.8) | 58.1, CH2 | 4.26, dd (13.5, 2.1) 4.02, dd (13.5, 4.5) |
| 14 | 123.3, CH | 6.18, d (16.0) | 123.8, CH | 6.04, d (16.1) | 120.6, CH | 6.04, m |
| 15 | 138.2, CH | 5.86, dd (16.0, 6.1) | 133.9, CH | 5.49, dt (16.1, 7.0) | 138.4, CH | 6.03, m |
| 16 | 71.3, CH | 3.94, m | 32.6, CH2 | 2.04, m | 33.0, CH2 | 2.10, m |
| 17 | 37.1, CH2 | 1.40, m 1.34, m | 28.4, CH2 | 1.32, m | 28.2, CH2 | 1.37, m |
| 18 | 27.2, CH2 | 1.27, m | 30.6, CH2 | 1.23, m | 30.7, CH2 | 1.26, m |
| 19 | 22.2, CH2 | 1.25, m | 21.9, CH2 | 1.27, m | 21.9, CH2 | 1.28, m |
| 20 | 14.0, CH3 | 0.86, t (6.9) | 13.9, CH3 | 0.86, t (6.8) | 13.9, CH3 | 0.86, t (7.0) |
| 3-OH | 5.07, d (4.9) | 5.12, d (4.9) | 5.26, d (5.0) | |||
| 7-OH | 4.91, d (7.3) | |||||
| 13-OH | 4.57, dd (5.0, 4.1) | 4.73, dd (5.8, 4.4) | 5.17, dd (4.5, 2.1) | |||
| 16-OH | 4.64, d (4.5) | |||||
| 7-OAc | 20.6, CH3 170.0, C | 2.02, s | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; Müller, W.E.G.; Meier, D.; Kalscheuer, R.; Guo, Z.; Zou, K.; Umeokoli, B.O.; Liu, Z.; Proksch, P. Polyketide Derivatives from Mangrove Derived Endophytic Fungus Pseudopestalotiopsis theae. Mar. Drugs 2020, 18, 129. https://doi.org/10.3390/md18020129
Yu X, Müller WEG, Meier D, Kalscheuer R, Guo Z, Zou K, Umeokoli BO, Liu Z, Proksch P. Polyketide Derivatives from Mangrove Derived Endophytic Fungus Pseudopestalotiopsis theae. Marine Drugs. 2020; 18(2):129. https://doi.org/10.3390/md18020129
Chicago/Turabian StyleYu, Xiaoqin, Werner E. G. Müller, Dieter Meier, Rainer Kalscheuer, Zhiyong Guo, Kun Zou, Blessing O. Umeokoli, Zhen Liu, and Peter Proksch. 2020. "Polyketide Derivatives from Mangrove Derived Endophytic Fungus Pseudopestalotiopsis theae" Marine Drugs 18, no. 2: 129. https://doi.org/10.3390/md18020129