Extraction Improvement of the Bioactive Blue-Green Pigment “Marennine” from Diatom Haslea ostrearia’s Blue Water: A Solid-Phase Method Based on Graphitic Matrices


Abstract
:1. Introduction
2. Material and Method
2.1. Algae Culture
2.2. Ultrafiltration and Dialysis: Previous Method Overview
2.3. Mobile Phase Optimization
2.3.1. Effect of Organic Modifier
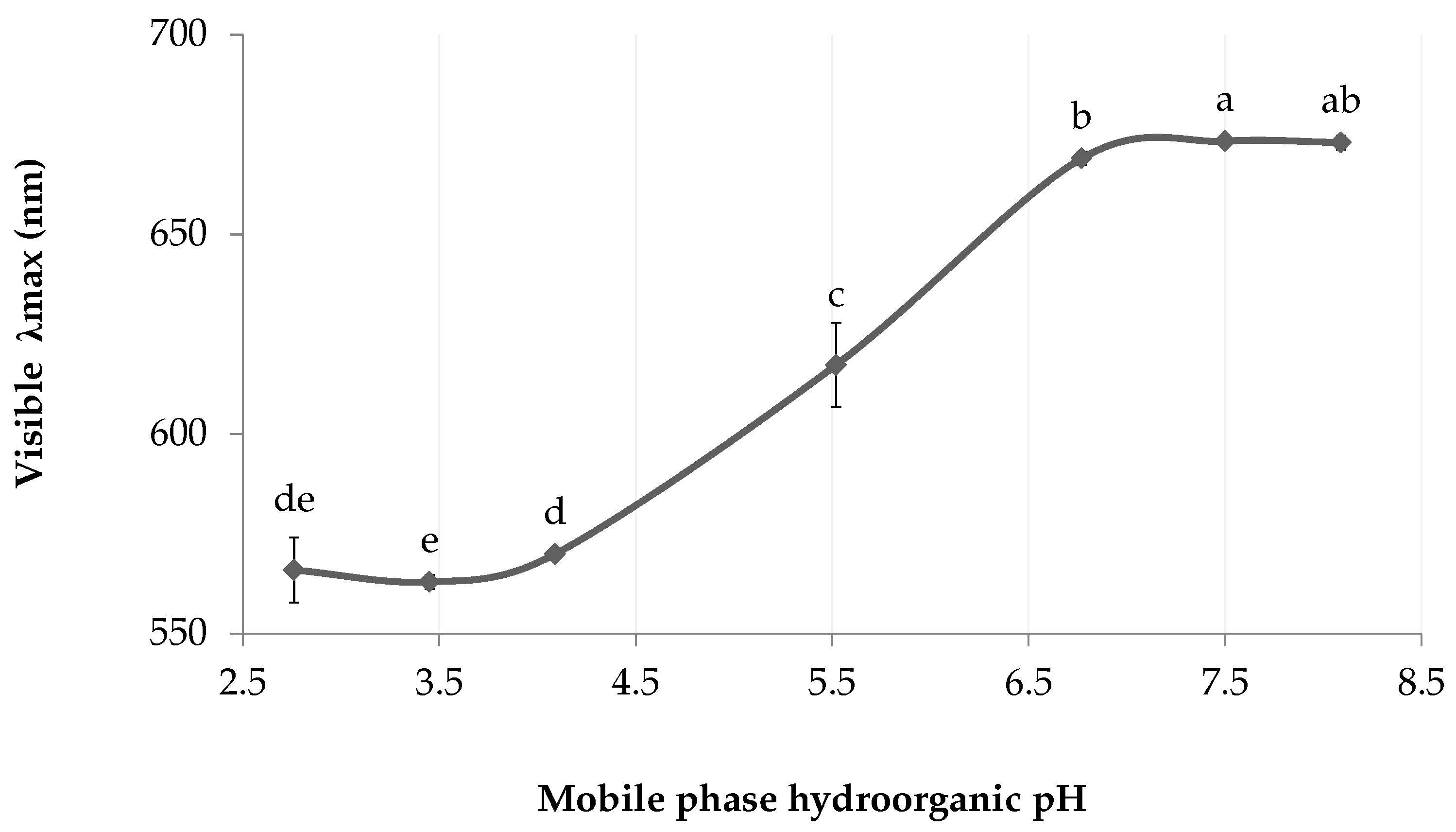
2.3.2. Effect of Analyte Ionization
2.3.3. Effect of Ionic Strength
2.3.4. Effect of Reductants
2.4. Stationary Phases Evaluation
2.4.1. Reductants and Repeatability
2.4.2. Evaluation of Graphite Flakes As a GCB Alternative
2.5. Crude Extract Recovery: Antisolvent Precipitation
2.6. Cations Removal
2.7. Purification
2.8. Instruments
2.9. Statistical Analysis
3. Results
3.1. Ultrafiltration Crude Yield: Previous Method
3.2. Overview of the Novel SPE-GCB Method
3.3. Effect of Organic Modifier
3.4. Effect of Analyte Ionization
3.5. Effect of Ionic Strength
3.6. Effect of Reductants
3.7. Stationary Phases Evaluation
3.7.1. Reductants and Repeatability
3.7.2. Evaluation of Graphite Flakes As GCB Alternative
3.8. Crude Extract Recovery: Antisolvent Precipitation
3.9. Cations Removal
3.10. Purification
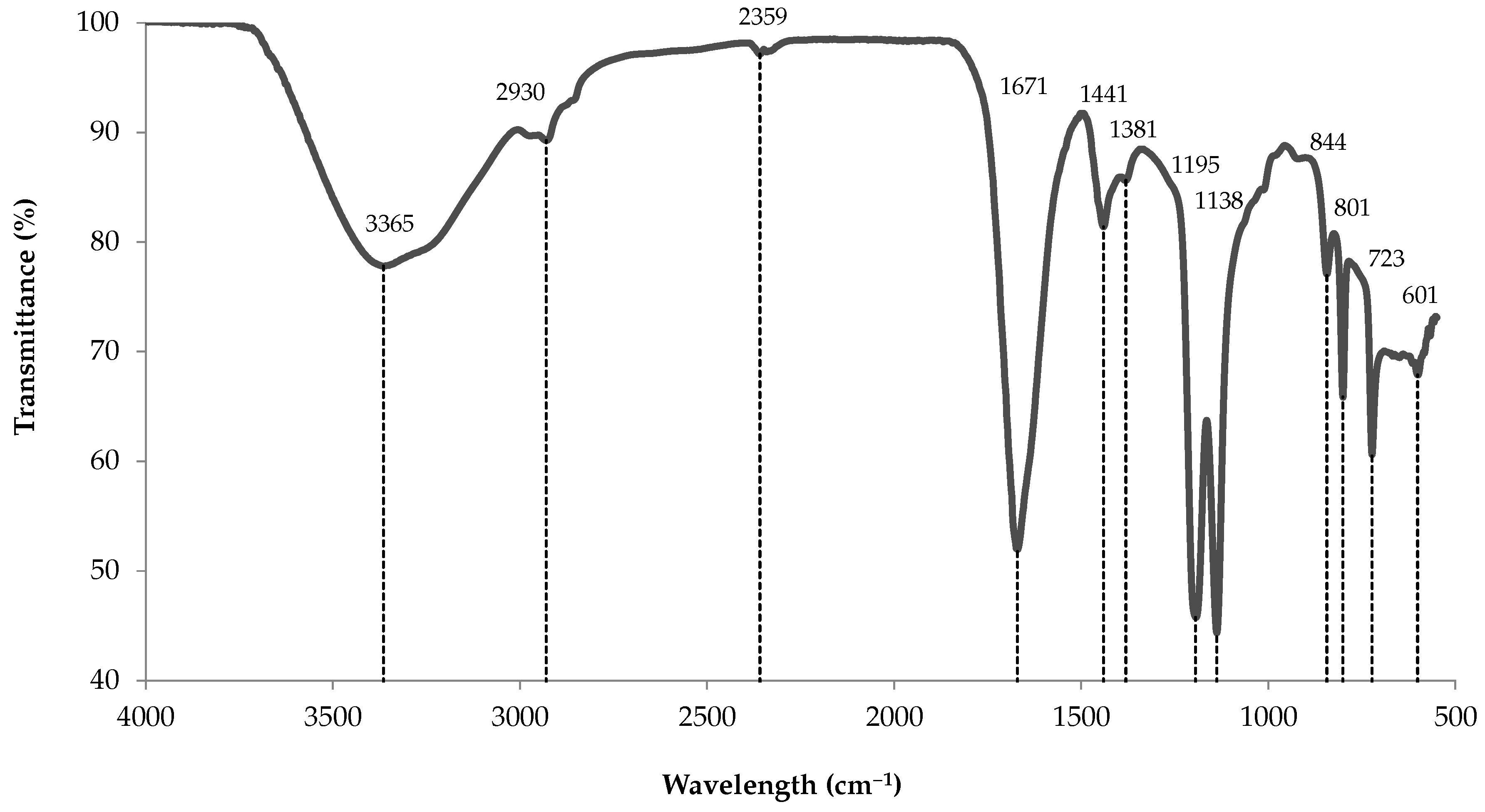
3.10.1. ATR-FTIR Analysis
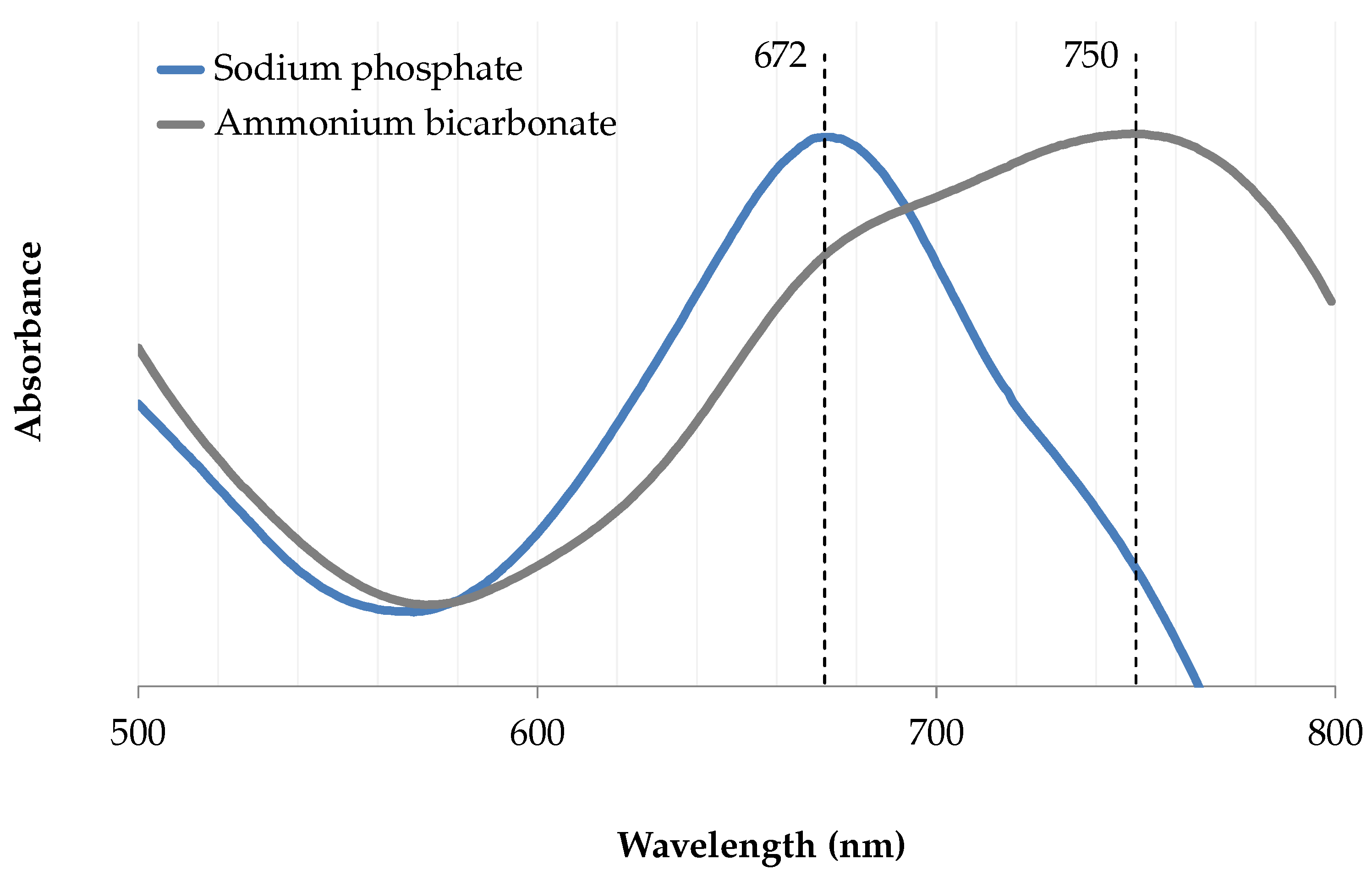
3.10.2. UV–Vis Analysis
3.10.3. NMR Analysis
4. Discussion
4.1. Optimization of Extraction Parameters
4.2. Extraction and Characterization of a Sulfated Polysaccharide
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATR | Attenuated total reflection |
| BW | Blue water (Haslea ostrearia culture supernatant) |
| DCM | Dichloromethane |
| DMSO | Dimethyl sulfoxide |
| EMn | Extracellular marennine |
| EPS | Exopolysaccharide |
| FTIR | Fourier-transform infrared spectroscopy |
| GCB | Graphitized carbon black |
| HPLC | High-performance liquid chromatography |
| HSQC | Heteronuclear single quantum correlation |
| IMn | Intracellular marennine |
| MP-AES | Microwave plasma-atomic emission spectrometer |
| MWCO | Molecular weight cutoff |
| MeOH | Methanol |
| NMR | Nuclear magnetic resonance spectroscopy |
| PBR | Photobioreactor |
| pH | Hydroorganic pH value of an electrode calibrated in aqueous buffers |
| pH | Aqueous pH value |
| PGC | Porous graphitic carbon |
| PTFE | Polytetrafluoroethylene |
| SCX | Strong cation exchange |
| SPE | Solid-phase extraction |
| TFA | Trifluoroacetic acid |
| UV–Vis | UV-Visible spectroscopy |
References
- Gastineau, R.; Turcotte, F.; Pouvreau, J.B.; Morançais, M.; Fleurence, J.; Windarto, E.; Prasetiya, F.S.; Arsad, S.; Jaouen, P.; Babin, M.; et al. Marennine, promising blue pigments from a widespread Haslea diatom species complex. Mar. Drugs 2014, 12, 3161–3189. [Google Scholar] [CrossRef] [Green Version]
- Pouvreau, J.B.; Morançais, M.; Fleury, F.; Rosa, P.; Thion, L.; Cahingt, B.; Zal, F.; Fleurence, J.; Pondaven, P. Preliminary characterisation of the blue-green pigment “marennine” from the marine tychopelagic diatom Haslea ostrearia (Gaillon/Bory) Simonsen. J. Appl. Phycol. 2006, 18, 757–767. [Google Scholar] [CrossRef]
- Falaise, C.; James, A.; Travers, M.A.; Zanella, M.; Badawi, M.; Mouget, J.L. Complex Relationships between the Blue Pigment Marennine and Marine Bacteria of the Genus Vibrio. Mar. Drugs 2019, 17, 160. [Google Scholar] [CrossRef] [Green Version]
- Turcotte, F.; Mouget, J.L.; Genard, B.; Lemarchand, K.; Deschênes, J.S.; Tremblay, R. Prophylactic effect of Haslea ostrearia culture supernatant containing the pigment marennine to stabilize bivalve hatchery production. Aquat. Living Resour. 2016, 29, 401. [Google Scholar] [CrossRef] [Green Version]
- Xuan, R.N.; Safitri, I.; Mouget, J.; Pruvost, J.; Turpin, V.; Jaouen, P. Design of an artificial culture medium to optimize Haslea ostrearia biomass and marennine production. Algal Res. 2020, 45, 101653. [Google Scholar] [CrossRef]
- Liška, I. Fifty years of solid-phase extraction in water analysis–historical development and overview. J. Chromatogr. A 2000, 885, 3–16. [Google Scholar] [CrossRef]
- Michel, M.; Buszewski, B. Porous graphitic carbon sorbents in biomedical and environmental applications. Adsorption 2009, 15, 193–202. [Google Scholar] [CrossRef]
- Shibukawa, M.; Terashima, H.; Nakajima, H.; Saitoh, K. Evaluation of the surface charge properties of porous graphitic carbon stationary phases treated with redox agents. Analyst 2004, 129, 623–628. [Google Scholar] [CrossRef]
- Guillard, R.R. Culture of phytoplankton for feeding marine invertebrates. In Culture of Marine Invertebrate Animals; Springer: Berlin/Heidelberg, Germany, 1975; pp. 29–60. [Google Scholar]
- Pouvreau, J.B.; Morançais, M.; Massé, G.; Rosa, P.; Robert, J.M.; Fleurence, J.; Pondaven, P. Purification of the blue-green pigment “marennine” from the marine tychopelagic diatom Haslea ostrearia (Gaillon/Bory) Simonsen. J. Appl. Phycol. 2006, 18, 769–781. [Google Scholar] [CrossRef]
- Wiczling, P.; Markuszewski, M.J.; Kaliszan, R. Determination of p K a by pH Gradient Reversed-Phase HPLC. Anal. Chem. 2004, 76, 3069–3077. [Google Scholar] [CrossRef] [PubMed]
- Tako, M.; Yoza, E.; Tohma, S. Chemical characterization of acetyl fucoidan and alginate from commercially cultured Cladosiphon okamuranus. Bot. Mar. 2000, 43, 393–398. [Google Scholar] [CrossRef]
- Berkhout, J.H.; Aswatha Ram, H. Recent Advancements in Spectrophotometric pKa Determinations: A Review. Indian J. Pharm. Educ. Res. 2019, 53, S475–S480. [Google Scholar] [CrossRef] [Green Version]
- Saboural, P.; Chaubet, F.; Rouzet, F.; Al-Shoukr, F.; Azzouna, R.B.; Bouchemal, N.; Picton, L.; Louedec, L.; Maire, M.; Rolland, L.; et al. Purification of a low molecular weight fucoidan for SPECT molecular imaging of myocardial infarction. Mar. Drugs 2014, 12, 4851–4867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziadi, M.; Bouzaiene, T.; M’Hir, S.; Zaafouri, K.; Mokhtar, F.; Hamdi, M.; Boisset-Helbert, C. Evaluation of the efficiency of ethanol precipitation and ultrafiltration on the purification and characteristics of exopolysaccharides produced by three lactic acid bacteria. BioMed Res. Int. 2018, 2018, 1896240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rani, V.; Shakila, R.; Jawahar, P.; Srinivasan, A. Influence of species, geographic location, seasonal variation and extraction method on the fucoidan yield of the brown seaweeds of Gulf of Mannar, India. Indian J. Pharm. Sci. 2017, 79, 65–71. [Google Scholar] [CrossRef]
- Pavia, D.L.; Lampman, G.M.; Kriz, G.S.; Vyvyan, J.A. Introduction to Spectroscopy, 5th ed.; Cengage Learning: Boston, MA, USA, 2013. [Google Scholar]
- Guo, Q.; Ai, L.; Cui, S. Methodology for Structural Analysis of Polysaccharides; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Ale, M.T.; Maruyama, H.; Tamauchi, H.; Mikkelsen, J.D.; Meyer, A.S. Fucose-containing sulfated polysaccharides from brown seaweeds inhibit proliferation of melanoma cells and induce apoptosis by activation of caspase-3 in vitro. Mar. Drugs 2011, 9, 2605–2621. [Google Scholar] [CrossRef] [Green Version]
- Pereira, L.; Amado, A.M.; Critchley, A.T.; Van de Velde, F.; Ribeiro-Claro, P.J. Identification of selected seaweed polysaccharides (phycocolloids) by vibrational spectroscopy (FTIR-ATR and FT-Raman). Food Hydrocoll. 2009, 23, 1903–1909. [Google Scholar] [CrossRef] [Green Version]
- Bilan, M.I.; Grachev, A.A.; Shashkov, A.S.; Nifantiev, N.E.; Usov, A.I. Structure of a fucoidan from the brown seaweed Fucus serratus L. Carbohydr. Res. 2006, 341, 238–245. [Google Scholar] [CrossRef]
- Jesumani, V.; Du, H.; Pei, P.; Aslam, M.; Huang, N. Comparative study on skin protection activity of polyphenol-rich extract and polysaccharide-rich extract from Sargassum vachellianum. PLoS ONE 2020, 15, e0227308. [Google Scholar] [CrossRef] [Green Version]
- Sichert, A.; Le Gall, S.; Klau, L.J.; Laillet, B.; Rogniaux, H.; Aachmann, F.L.; Hehemann, J.H. Ion-exchange purification and structural characterization of five sulfated fucoidans from brown algae. Glycobiology 2020. [Google Scholar] [CrossRef]
- Sichert, A. Fucoidan Degradation by Marine Bacteria. Ph.D. Thesis, Universität Bremen, Bremen, Germany, 2020. [Google Scholar]
- Bilan, M.I.; Grachev, A.A.; Ustuzhanina, N.E.; Shashkov, A.S.; Nifantiev, N.E.; Usov, A.I. Structure of a fucoidan from the brown seaweed Fucus evanescens C. Ag. Carbohydr. Res. 2002, 337, 719–730. [Google Scholar] [CrossRef]
- Gügi, B.; Le Costaouec, T.; Burel, C.; Lerouge, P.; Helbert, W.; Bardor, M. Diatom-specific oligosaccharide and polysaccharide structures help to unravel biosynthetic capabilities in diatoms. Mar. Drugs 2015, 13, 5993–6018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhang, Q.; Zhang, Z.; Song, H.; Li, P. Potential antioxidant and anticoagulant capacity of low molecular weight fucoidan fractions extracted from Laminaria japonica. Int. J. Biol. Macromol. 2010, 46, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L. Therapeutic and Nutritional Uses of Algae; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Pierre, G.; Sopena, V.; Juin, C.; Mastouri, A.; Graber, M.; Maugard, T. Antibacterial activity of a sulfated galactan extracted from the marine alga Chaetomorpha aerea against Staphylococcus aureus. Biotechnol. Bioprocess Eng. 2011, 16, 937–945. [Google Scholar] [CrossRef]
- Rao, E.V.; Ramana, K.S. Structural studies of a polysaccharide isolated from the green seaweed Chaetomorpha anteninna. Carbohydr. Res. 1991, 217, 163–170. [Google Scholar]
- Monser, L. Liquid chromatographic determination of four purine bases using porous graphitic carbon column. Chromatographia 2004, 59, 455–459. [Google Scholar] [CrossRef]
- Barrett, D.; Pawula, M.; Knaggs, R.; Shaw, P. Retention behavior of morphine and its metabolites on a porous graphitic carbon column. Chromatographia 1998, 47, 667–672. [Google Scholar] [CrossRef]
- Reijenga, J.; van Hoof, A.; van Loon, A.; Teunissen, B. Development of methods for the determination of pKa values. Anal. Chem. Insights 2013, 8, ACI–S12304. [Google Scholar] [CrossRef] [Green Version]
- Karaffa, L.S. The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals; RSC Publishing: Cambridge, UK, 2013. [Google Scholar]
- Bapiro, T.E.; Richards, F.M.; Jodrell, D.I. Understanding the complexity of porous graphitic carbon (PGC) chromatography: Modulation of mobile-stationary phase interactions overcomes loss of retention and reduces variability. Anal. Chem. 2016, 88, 6190–6194. [Google Scholar] [CrossRef] [Green Version]
- West, C.; Elfakir, C.; Lafosse, M. Porous graphitic carbon: A versatile stationary phase for liquid chromatography. J. Chromatogr. A 2010, 1217, 3201–3216. [Google Scholar] [CrossRef]
- Chemat, F.; Vian, M.A. Alternative Solvents for Natural Products Extraction; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Capello, C.; Fischer, U.; Hungerbühler, K. What is a green solvent? A comprehensive framework for the environmental assessment of solvents. Green Chem. 2007, 9, 927–934. [Google Scholar] [CrossRef]
- Tobiszewski, M.; Namieśnik, J. Greener organic solvents in analytical chemistry. Curr. Opin. Green Sustain. Chem. 2017, 5, 1–4. [Google Scholar] [CrossRef]
- Golden, J.S.; Handfield, R.B. Why biobased? In Opportunitiesin the Emerging Bioeconomy; US Department of Agriculture: Washington, DC, USA, 2014; Volume 40. [Google Scholar]
- Kurian, J.K.; Nair, G.R.; Hussain, A.; Raghavan, G.V. Feedstocks, logistics and pre-treatment processes for sustainable lignocellulosic biorefineries: A comprehensive review. Renew. Sustain. Energy Rev. 2013, 25, 205–219. [Google Scholar] [CrossRef]
- Pereira, C.S.; Silva, V.M.; Rodrigues, A.E. Ethyl lactate as a solvent: Properties, applications and production processes—A review. Green Chem. 2011, 13, 2658–2671. [Google Scholar] [CrossRef]
- Micăle, F.; Albu, F.; Iorgulescu, E.E.; Medvedovici, A.; Tache, F. Ethyl Lactate as a Greener Alternative to Acetonitrile in RPLC: A Realistic Appraisal. J. Chromatogr. Sci. 2015, 53, 1701–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennion, M.C. Graphitized carbons for solid-phase extraction. J. Chromatogr. A 2000, 885, 73–95. [Google Scholar] [CrossRef]
- Ruhaak, L.R.; Deelder, A.M.; Wuhrer, M. Oligosaccharide analysis by graphitized carbon liquid chromatography–mass spectrometry. Anal. Bioanal. Chem. 2009, 394, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Funari, C.S.; Carneiro, R.L.; Khandagale, M.M.; Cavalheiro, A.J.; Hilder, E.F. Acetone as a greener alternative to acetonitrile in liquid chromatographic fingerprinting. J. Sep. Sci. 2015, 38, 1458–1465. [Google Scholar] [CrossRef]
- Smallwood, I.M. Solvent Recovery Handbook, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2002. [Google Scholar]
- Miller, R.L.; Guimond, S.E.; Prescott, M.; Turnbull, J.E.; Karlsson, N. Versatile separation and analysis of heparan sulfate oligosaccharides using graphitized carbon liquid chromatography and electrospray mass spectrometry. Anal. Chem. 2017, 89, 8942–8950. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Kojima, T.; Miwa, T. Ion chromatography of inorganic anions on graphitic carbon as the stationary phase. J. High Resolut. Chromatogr. 2000, 23, 590–594. [Google Scholar] [CrossRef]
- Shibukawa, M.; Unno, A.; Oyashiki, Y.; Nagoya, A.; Oguma, K.; Miura, T. Redox reaction catalyzed by a porous graphite carbon packing and its application to selectivity enhancement of high-performance liquid chromatography separation of metal complexes. Anal. Commun. 1997, 34, 397–400. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, Q.; Yan, X.; Li, H.; Zhang, P.; Wang, L.; Zhou, T.; Li, Y.; Ding, L. Rapid preparation of expanded graphite by microwave irradiation for the extraction of triazine herbicides in milk samples. Food Chem. 2016, 197, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Kerton, F.M.; Marriott, R. Alternative Solvents for Green Chemistry; Number 20; Royal Society of Chemistry: London, UK, 2013. [Google Scholar]
- Pouvreau, J.B. Purification et Caractérisation du Pigment Bleu-Vert “Marennine” Synthétisé par la Diatomée Marine Haslea ostrearia (Gaillon/Bory) Simonsen: Propriétés Physico-Chimiques et Activités Biologiques. Ph.D. Thesis, Université de Nantes, Nantes, France, 2006. [Google Scholar]
- Li, B.; Lu, F.; Wei, X.; Zhao, R. Fucoidan: Structure and bioactivity. Molecules 2008, 13, 1671–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, P.; Yin, Z.; Qu, G.; Wang, C. Fucoidan and Its Health Benefits. In Bioactive Seaweeds for Food Applications; Elsevier: Amsterdam, The Netherlands, 2018; pp. 223–238. [Google Scholar]
- Ale, M.T.; Meyer, A.S. Fucoidans from brown seaweeds: An update on structures, extraction techniques and use of enzymes as tools for structural elucidation. RSC Adv. 2013, 3, 8131–8141. [Google Scholar] [CrossRef] [Green Version]
- Gogou, A.; Repeta, D.J. Particulate-dissolved transformations as a sink for semi-labile dissolved organic matter: Chemical characterization of high molecular weight dissolved and surface-active organic matter in seawater and in diatom cultures. Mar. Chem. 2010, 121, 215–223. [Google Scholar] [CrossRef]
- Satpati, G.G. Algal Sulfated Polysaccharides: Potent Immunomodulators against COVID-19 in Pandemic 2020. Biosci. Biotechnol. Res. Asia 2020, 17, 601–605. [Google Scholar] [CrossRef]
- Song, S.; Peng, H.; Wang, Q.; Liu, Z.; Dong, X.; Wen, C.; Ai, C.; Zhang, Y.; Wang, Z.; Zhu, B.W. Inhibitory activities of marine sulfated polysaccharides against SARS-CoV-2. Food Funct. 2020, 11, 7415–7420. [Google Scholar] [CrossRef]
- Kwon, P.S.; Oh, H.; Kwon, S.J.; Jin, W.; Zhang, F.; Fraser, K.; Hong, J.J.; Linhardt, R.J.; Dordick, J.S. Sulfated polysaccharides effectively inhibit SARS-CoV-2 in vitro. Cell Discov. 2020, 6, 50. [Google Scholar] [CrossRef]
- Nishiguchi, T.; Jiang, Z.; Ueno, M.; Takeshita, S.; Cho, K.; Roh, S.W.; Kang, K.H.; Yamaguchi, K.; Kim, D.; Oda, T. Reevaluation of bactericidal, cytotoxic, and macrophage-stimulating activities of commercially available Fucus vesiculosus fucoidan. Algae 2014, 29, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Holtkamp, A.D.; Kelly, S.; Ulber, R.; Lang, S. Fucoidans and fucoidanases—Focus on techniques for molecular structure elucidation and modification of marine polysaccharides. Appl. Microbiol. Biotechnol. 2009, 82, 1. [Google Scholar] [CrossRef]
- Guo, M.Q.; Hu, X.; Wang, C.; Ai, L. Polysaccharides: Structure and solubility. In Solubility of Polysaccharides; BoD—Books on Demand: Norderstedt, Germany, 2017; pp. 7–21. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lot | BW Concentration (mgL) | Ultrafiltration Yield (Non-Dialyzed, %) | Ultrafiltration Yield (Reference, %) [10] |
|---|---|---|---|
| 1 | 9.3 | 54.9 | – |
| 2 | 11.7 | 55.3 | – |
| 3 | 12.3 | 59.5 | – |
| Average | 11.1 (1.6) | 56.6 (2.6) | 62.5 |
| Fractions | 1 | 5–10 | 11–20 | Average |
|---|---|---|---|---|
| Reductants | 61 (3) | 66 (2) | 67 (2) | 66 (3) |
| Blank | 47 (3) | 66 (2) | 67 (2) | 64 (5) |
| Stationary Phase | Saturation (AUg) | Yield (%) |
|---|---|---|
| Graphitized carbon black (GCB) | 23.03 (0.97) (a) | 64.90 (4.04) (a) |
| Graphite flakes | 15.69 (0.59) (b) | 61.97 (1.40) (a) |
| Purification Step | Yield (%) | Na+ | K+ | Mg2+ | Ca2+ |
|---|---|---|---|---|---|
| Blue water | 100 | 10,095 (56) | 348 (8) | 1109 (13) | 372 (5) |
| Precipitation | 49.5 (1.0) | 1609 (32) | 5.18 (0.51) | 7.10 (0.35) | 4.17 (0.08) |
| Pellet rinse | 48.4 (0.3) | 1596 (105) | 4.30 (1.30) | 7.29 (0.05) | 3.20 (0.04) |
| Desalting (SCX) | 36.5 (3.9) | 51 (89) | 0.10 (0.07) | 0.24 (0.13) | 0.63 (0.48) |
| Experimental Wavelength (cm) | Fucoidan Wavelength (cm) | Assignment | Reference |
|---|---|---|---|
| 3365 (4) | 3448 | Hydrogen bonded O–H broad band | [16,17] |
| 2930 (1) | 2940 | C–H stretching of pyranose ring | [14] |
| 1671 (4) | 1688 | O–C–O asymmetric stretching | [16] |
| 1441 (0) | 1437 | O–C–O symmetric stretching | [16,18] |
| 1381 (2) | – | C–H bending | [18] |
| 1195 (7) | 1203 | C–H deformation of -manuronic residues | [16] |
| 1138 (2) | 1140 | O=S=O symmetric stretching | [16,17] |
| 844 (0) | 844 | S=O stretching | [16] |
| 801 (1) | 803 | Sulphate group absorption band | [16] |
| 723 (1) | 724 | C–O–S stretching | [16] |
| 601 (1) | 601 | C=C–H stretching | [16] |
| Sample | Extraction Method | 247/322 | 247/677 |
|---|---|---|---|
| Polysaccharides fraction | SPE-GCB | 2.53 (0.02) | – |
| Marennine fraction | SPE-GCB | 2.60 (0.04) | 5.07 (0.13) |
| Ultrafiltrated marennine | 3–30 kDa | 2.86 (0.03) | 5.49 (0.07) |
| Purified reference [2] | 3–30 kDa, anion-exchange | 2.63 | 3.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bélanger, W.; Arnold, A.A.; Turcotte, F.; Saint-Louis, R.; Deschênes, J.-S.; Genard, B.; Marcotte, I.; Tremblay, R. Extraction Improvement of the Bioactive Blue-Green Pigment “Marennine” from Diatom Haslea ostrearia’s Blue Water: A Solid-Phase Method Based on Graphitic Matrices. Mar. Drugs 2020, 18, 653. https://doi.org/10.3390/md18120653
Bélanger W, Arnold AA, Turcotte F, Saint-Louis R, Deschênes J-S, Genard B, Marcotte I, Tremblay R. Extraction Improvement of the Bioactive Blue-Green Pigment “Marennine” from Diatom Haslea ostrearia’s Blue Water: A Solid-Phase Method Based on Graphitic Matrices. Marine Drugs. 2020; 18(12):653. https://doi.org/10.3390/md18120653
Chicago/Turabian StyleBélanger, William, Alexandre A. Arnold, François Turcotte, Richard Saint-Louis, Jean-Sébastien Deschênes, Bertrand Genard, Isabelle Marcotte, and Réjean Tremblay. 2020. "Extraction Improvement of the Bioactive Blue-Green Pigment “Marennine” from Diatom Haslea ostrearia’s Blue Water: A Solid-Phase Method Based on Graphitic Matrices" Marine Drugs 18, no. 12: 653. https://doi.org/10.3390/md18120653