Synthetic Approaches to Zetekitoxin AB, a Potent Voltage-Gated Sodium Channel Inhibitor


Abstract
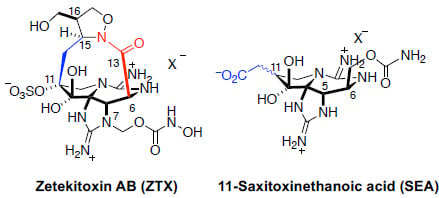
:
1. Introduction
1.1. Voltage-Gated Sodium Channel Isoforms
1.2. Saxitoxin As A NaV Modulator
1.3. Natural Analogs of Saxitoxin, Including Zetekitoxin AB (ZTX)
1.4. Scope of This Review
2. Development of Carbon–Carbon Linkage at C11 of STX, And Application to The Synthesis of 11-Saxitoxinethanoic Acid (SEA, 9)
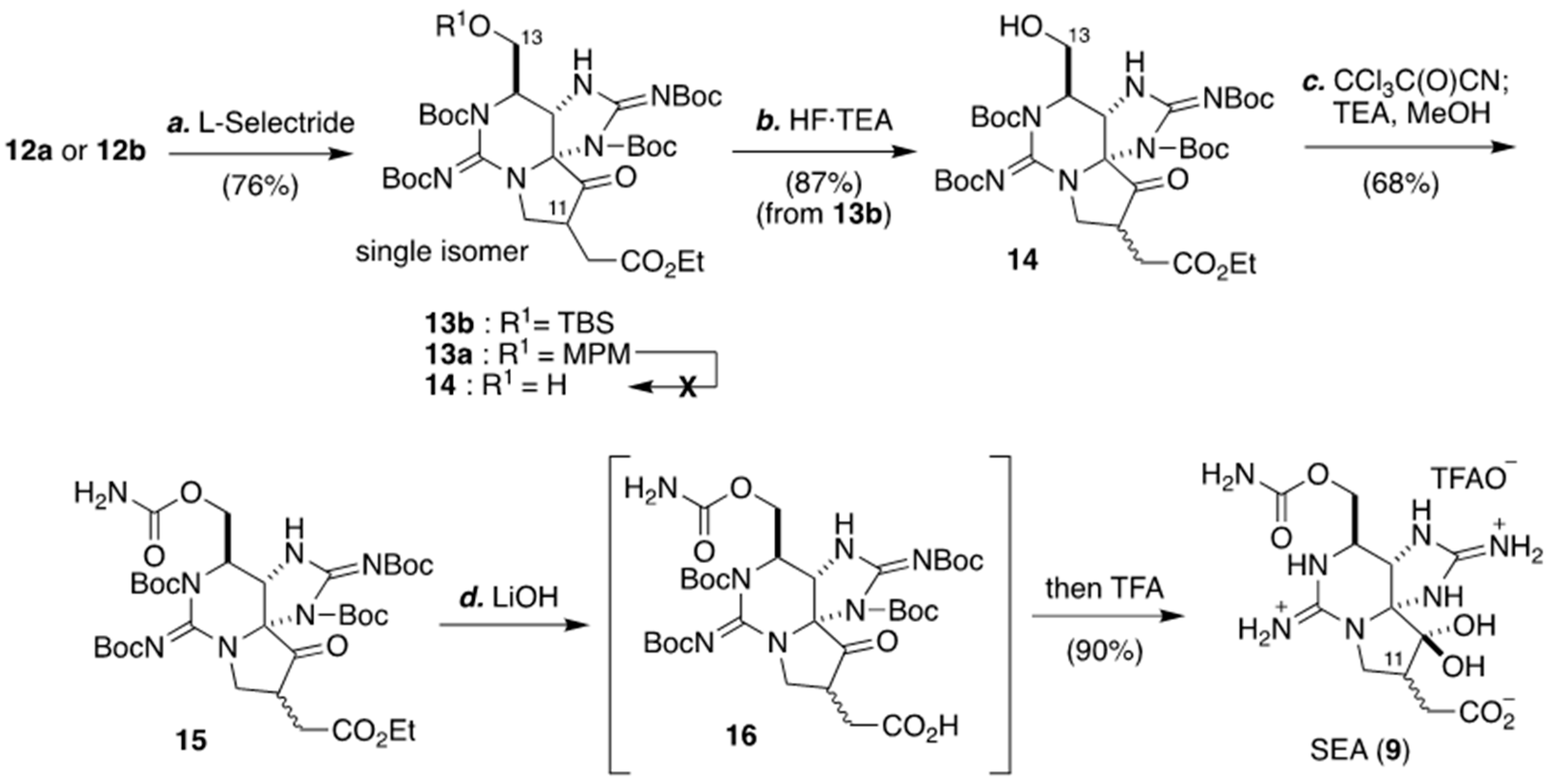
2.1. Carbon–Carbon Bond Formation at C11 by Mukaiyama Aldol Condensation Reaction, as Applied for The Synthesis of (+)-SEA by Nagasawa’s Group
2.2. Carbon–Carbon Bond Formation At C11 by Stille Coupling Reaction, As Applied for The Synthesis of (+)-SEA by Du Bois’ Group
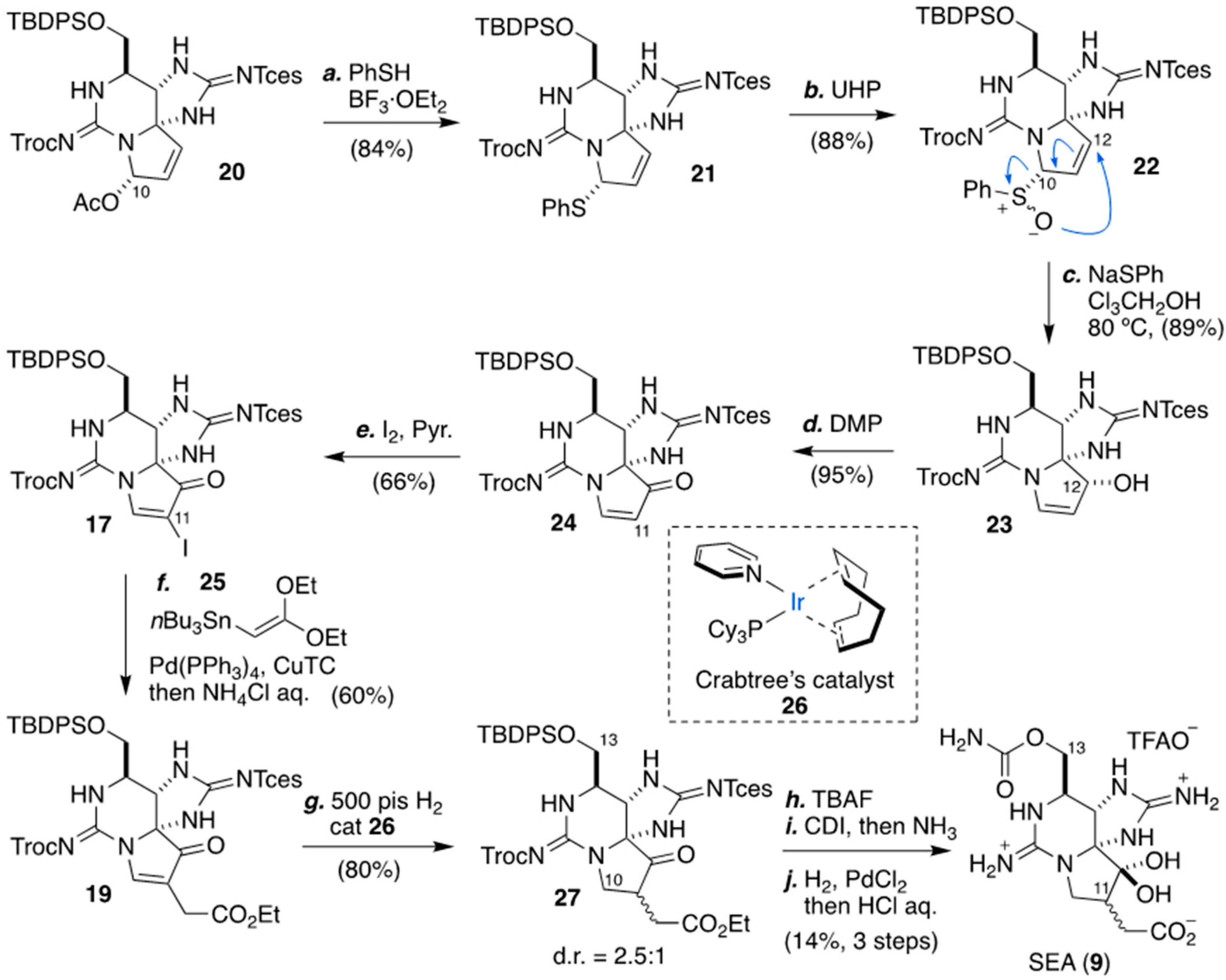
2.3. Carbon–Carbon Bond Formation at C11 by C-Alkylation, As Applied for The Synthesis of (+)-SEA by Looper’s Group
2.4. NaV-Inhibitory Activity of Synthesized, C11-Substituted Saxitoxin Analogs
3. Stereoselective Synthesis of The Isoxazolidine Moiety of ZTX (8), And Its Introduction at C13 in A Model Compound
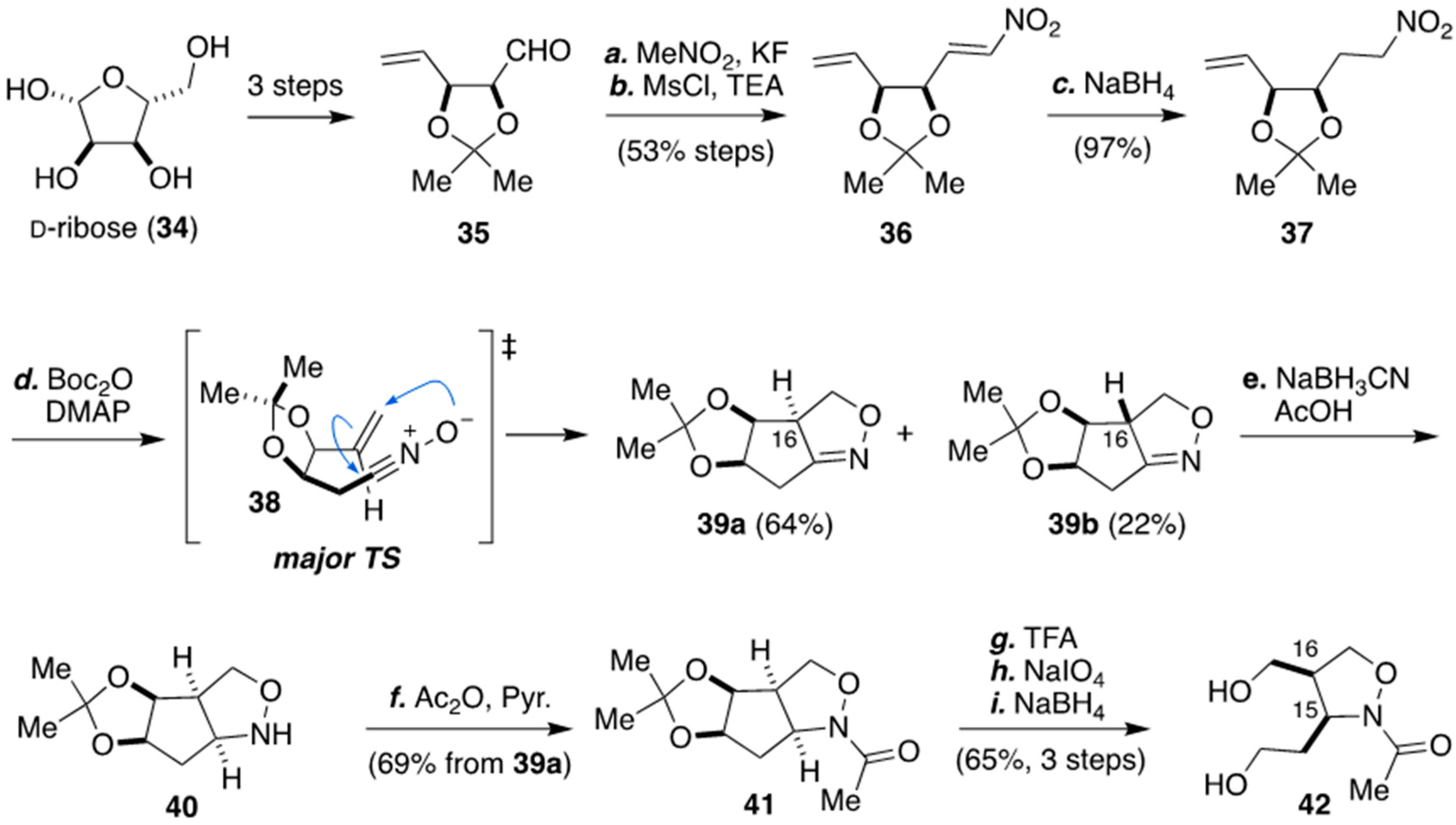
3.1. Synthesis of The Isoxazolidine Part of Zetekitoxin (8) from D-ribose by Nishikawa And Co-workers
3.2. Stereoselective Synthesis of The Isoxazolidine Part from Methyl α-d-glucopyranoside by Lopper and Co-Workers
3.3. Comparison of the Chemical Shift at C13 in Zetekitoxin (8) with Those in Some Synthetic Models
4. Synthesis of The Characteristic Macrocyclic Structure of ZTX (8) by Looper’s Group
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hodgkin, A.L.; Huxley, A.F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952, 117, 500–544. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.L.; Barchi, R.L.; Caldwell, J.H.; Hofmann, F.; Howe, J.R.; Hunter, J.C.; Kallen, R.G.; Mandel, G.; Meisler, M.H.; Netter, Y.B.; et al. Nomenclature of voltage-gated sodium channels. Neuron 2000, 28, 365–368. [Google Scholar] [CrossRef] [Green Version]
- Narahashi, T. Tetrodotoxin. Proc. Jpn. Acad. Ser. B 2008, 84, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, T.; Isobe, M. Synthesis of Tetrodotoxin, a Classic but Still Fascinating Natural Product. Chem. Rec. 2013, 13, 286–302. [Google Scholar] [CrossRef]
- Moczydlowski, E.G. The molecular mystique of tetrodotoxin. Toxicon 2013, 63, 165–183. [Google Scholar] [CrossRef]
- Fozzard, H.A.; Lipkind, G.M. The Tetrodotoxin Binding Site Is within the Outer Vestibule of the Sodium Channel. Mar. Drugs 2010, 8, 219–234. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Ruben, P.C. Interaction between voltage-gated sodium channels and the neurotoxin, tetrodotoxin. Channels 2008, 2, 407–412. [Google Scholar] [CrossRef]
- Clare, J.J.; Tate, S.N.; Nobbs, M.; Romanos, M.A. Voltage-gated sodium channels as therapeutic targets. Drug Discov. Today 2000, 5, 506–520. [Google Scholar] [CrossRef]
- Noda, M.; Ikeda, T.; Kayano, T.; Suzuki, H.; Takeshima, H.; Kurasaki, M.; Takahashi, H.; Numa, S. Existence of distinct sodium channel messenger RNAs in rat brain. Nature 1986, 320, 188–192. [Google Scholar] [CrossRef]
- Meadows, L.S.; Chen, Y.H.; Powell, A.J.; Clare, J.J.; Ragsdale, D.S. Functional modulation of human brain NaV1.3 sodium channels, expressed in mammalian cells, by auxiliary beta 1, beta 2 and beta 3 subunits. Neuroscience 2002, 114, 745–753. [Google Scholar] [CrossRef]
- Trimmer, J.S.; Cooperman, S.S.; Tomiko, S.A.; Zhou, J.; Crean, S.M.; Boyle, M.B.; Kallen, R.G.; Sheng, Z.; Barchi, R.L.; Sigworth, F.J. Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron 1989, 3, 33–49. [Google Scholar] [CrossRef]
- Chahine, M.; Bennett, P.B.; George, A.L., Jr.; Horn, R. Functional expression and properties of the human skeletal muscle sodium channel. Pflug. Arch. Eur. J. Physiol. 1994, 427, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, P.S.; McGivern, J.G.; Delgado, S.G.; Koch, B.D.; Eglen, R.M.; Hunter, J.C.; Sangameswaran, L. Functional analysis of a voltage-gated sodium channel and its splice variant from rat dorsal root ganglia. J. Neurochem. 1998, 70, 2262–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.R.; Smith, R.D.; Plummer, N.W.; Meisler, M.H.; Goldin, A.L. Functional Analysis of the Mouse Scn8a Sodium Channel. J. Neurosci. 1998, 18, 6093–6102. [Google Scholar] [CrossRef] [PubMed]
- Klugbauer, N.; Lacinova, L.; Flockerzi, V.; Hofmann, F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 1995, 14, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.R.; Howe, J.R.; Waxman, S.G. Slow Closed-State Inactivation: A Novel Mechanism Underlying Ramp Currents in Cells Expressing the hNE/PN1 Sodium Channel. J. Neurosci. 1998, 18, 9607–9619. [Google Scholar] [CrossRef]
- Sangameswaran, L.; Fish, L.M.; Koch, B.D.; Rabert, D.K.; Delgado, S.G.; Ilnicka, M.; Jakeman, L.B.; Novakovic, S.; Wong, K.; Sze, P.; et al. A Novel Tetrodotoxin-sensitive, Voltage-gated Sodium Channel Expressed in Rat and Human Dorsal Root Ganglia. J. Biol. Chem. 1997, 272, 14805–14809. [Google Scholar] [CrossRef] [Green Version]
- Santarelli, V.P.; Eastwood, A.L.; Dougherty, D.A.; Horn, R.; Ahern, C.A. A cation-pi interaction discriminates among sodium channels that are either sensitive or resistant to tetrodotoxin block. J. Biol. Chem. 2007, 282, 8044–8051. [Google Scholar] [CrossRef] [Green Version]
- Satin, J.; Kyle, J.W.; Chen, M.; Bell, P.; Cribbs, L.L.; Fozzard, H.A.; Rogart, R.B. A Mutant of TTX-Resistant Cardiac Sodium Channels with TTX-Sensitive Properties. Science 1992, 256, 1202–1205. [Google Scholar] [CrossRef]
- Sangameswaran, L.; Delgado, S.G.; Fish, L.M.; Koch, B.D.; Jakeman, L.B.; Stewart, G.R.; Sze, P.; Hunter, J.C.; Eglen, R.M.; Herman, R.C. Structure and Function of a Novel Voltage-gated, Tetrodotoxin-resistant Sodium Channel Specific to Sensory Neurons. J. Biol. Chem. 1996, 271, 5953–5956. [Google Scholar] [CrossRef] [Green Version]
- Akopian, A.N.; Sivilotti, L.; Wood, J.N. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature 1996, 379, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Tyrrell, L.; Black, J.A.; Waxman, S.G. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc. Natl. Acad. Sci. USA 1998, 95, 8963–8968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dib-Hajj, S.; Black, J.A.; Cummins, T.R.; Waxman, S.G. NaN/NaV1.9: A sodium channel with unique properties. Trends Neurosci. 2002, 25, 253–259. [Google Scholar] [CrossRef]
- McKerrall, S.J.; Sutherlin, D.P. NaV1.7 inhibitors for the treatment of chronic pain. Bioorg. Med. Chem. Lett. 2018, 28, 3141–3149. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, J.V.; Pajouhesh, H.; Beckley, J.T.; Delwig, A.; Du Bois, J.; Hunter, J.C. Challenges and Opportunities for Therapeutics Targeting the Voltage-Gated Sodium Channel Isoform NaV1.7. J. Med. Chem. 2019, 62, 8695–8710. [Google Scholar] [CrossRef] [PubMed]
- Bagal, S.K.; Kemp, M.L.; Bungay, P.J.; Hay, T.L.; Murata, Y.; Payne, C.E.; Stevens, E.B.; Brown, A.; Blakemore, D.C.; Corbett, M.S.; et al. Discovery and optimisation of potent and highly subtype selective NaV1.8 inhibitors with reduced cardiovascular liabilities. Med. Chem. Comm. 2016, 7, 1925–1931. [Google Scholar] [CrossRef]
- Kort, M.E.; Atkinson, R.N.; Thomas, J.B.; Drizin, I.; Johnson, M.S.; Secrest, M.A.; Gregg, R.J.; Scanio, M.J.; Shi, L.; Hakeem, A.H.; et al. Subtype-selective NaV1.8 sodium channel blockers: Identification of potent, orally active nicotinamide derivatives. Bioorg. Med. Chem. Let. 2010, 20, 6812–6815. [Google Scholar] [CrossRef]
- Jarvis, M.F.; Honore, P.; Shieh, C.C.; Chapman, M.; Joshi, S.; Zhang, X.F.; Kort, M.; Carroll, W.; Marron, B.; Atkinson, R.; et al. A-803467, a potent and selective NaV1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. USA 2007, 104, 8520–8525. [Google Scholar] [CrossRef] [Green Version]
- Kort, M.E.; Drizin, I.; Gregg, R.J.; Scanio, M.J.; Shi, L.; Gross, M.F.; Atkinson, R.N.; Johnson, M.S.; Pacofsky, G.J.; Thomas, J.B.; et al. Discovery and biological evaluation of 5-aryl-2-furfuramides, potent and selective blockers of the NaV1.8 sodium channel with efficacy in models of neuropathic and inflammatory pain. J. Med. Chem. 2008, 51, 407–416. [Google Scholar] [CrossRef]
- Priest, B.T.; Kaczorowski, G.J. Subtype-selective sodium channel blockers promise a new era of pain research. Proc. Natl. Acad. Sci. USA 2007, 104, 8205–8206. [Google Scholar] [CrossRef] [Green Version]
- England, S.; de Groot, M.J. Subtype-selective targeting of voltage-gated sodium channels. Br. J. Pharm. 2009, 158, 1413–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llewellyn, L.E. Saxitoxin, a toxic marine natural product that targets a multitude of receptors. Nat. Prod. Rep. 2006, 23, 200–222. [Google Scholar] [CrossRef] [PubMed]
- Cusick, K.D.; Sayler, G.S. An Overview on the Marine Neurotoxin, Saxitoxin: Genetics, Molecular Targets, Methods of Detection and Ecological Functions. Mar. Drugs 2013, 11, 991–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, H.; Meyer, G.F. Paralytic shellfish poisoning. Arch. Pathol. 1937, 24, 560–598. [Google Scholar]
- Sommer, H.; Whedon, W.F.; Kofoid, C.A.; Strohler, R. Relation of paralytic shellfish poison to certain plankton organisms of the genus Gonyaulax. Arch. Pathol. 1937, 24, 537–559. [Google Scholar]
- Schantz, E.J.; Mold, J.D.; Stanger, D.W.; Shavel, J.; Riel, F.J.; Bowden, J.P.; Lynch, J.M.; Wyler, R.S.; Riegel, B.; Sommer, H. Paralytic Shellfish Poison. VI. A Procedure for the Isolation and Purification of the Poison from Toxic Clam and Mussel Tissues. J. Am. Chem. Soc. 1957, 79, 5230–5235. [Google Scholar] [CrossRef]
- Mold, J.D.; Bowden, J.P.; Stanger, D.W.; Maurer, J.E.; Lynch, J.M.; Wyler, R.S.; Schantz, E.J.; Riegel, B. Paralytic Shellfish Poison. VII. Evidence for the Purity of the Poison Isolated from Toxic Clams and Mussels. J. Am. Chem. Soc. 1957, 79, 5235–5238. [Google Scholar] [CrossRef]
- Schuett, W.; Rapoport, H. Saxitoxin, the Paralytic Shellfish Poison. Degradation to a Pyrrolopyrimidine. J. Am. Chem. Soc. 1962, 84, 2266–2267. [Google Scholar] [CrossRef]
- Russell, F.E. Comparative pharmacology of some animal toxins. Fed. Proc. 1967, 26, 1206–1224. [Google Scholar]
- Bordner, J.; Thiessen, W.E.; Bates, H.A.; Rapoport, H. Structure of a crystalline derivative of saxitoxin. Structure of saxitoxin. J. Am. Chem. Soc. 1975, 97, 6008–6012. [Google Scholar] [CrossRef]
- Rogers, P.S.; Rapoport, H. The pKa’s of saxitoxin. J. Am. Chem. Soc. 1980, 102, 7335–7339. [Google Scholar] [CrossRef]
- Henderson, R.; Ritchie, J.M.; Strichartz, G.R. Evidence that tetrodotoxin and saxitoxin act at a metal cation binding site in the sodium channels of nerve membrane. Proc. Natl. Acad. Sci. USA 1974, 71, 3936–3940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hille, B. The receptor for tetrodotoxin and saxitoxin. A structural hypothesis. Biophys. J. 1975, 15, 615–619. [Google Scholar] [PubMed] [Green Version]
- Pan, X.; Li, Z.; Zhou, Q.; Shen, H.; Wu, K.; Huang, X.; Chen, J.; Zhang, J.; Zhu, X.; Lei, J.; et al. Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science 2018, 362, eaau2596. [Google Scholar]
- Shen, H.; Liu, D.; Wu, K.; Lei, J.; Yan, N. Structures of human NaV1.7 channel in complex with auxiliary subunits and animal toxins. Science 2019, 363, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.R.; Novick, P.A.; Parsons, W.H.; McGregor, M.; Zablocki, J.; Pande, V.S.; Du Bois, J. Marked difference in saxitoxin and tetrodotoxin affinity for the human nociceptive voltage-gated sodium channel (NaV1.7). Proc. Natl. Acad. Sci. USA 2012, 109, 18102–18107. [Google Scholar] [CrossRef] [Green Version]
- Thomas-Tran, R.; Du Bois, J. Mutant cycle analysis with modified saxitoxins reveals specific interactions critical to attaining high-affinity inhibition of hNaV1.7. Proc. Natl. Acad. Sci. USA 2016, 113, 5856–5861. [Google Scholar] [CrossRef] [Green Version]
- Wiese, M.; D’Agostino, P.M.; Mihali, T.K.; Moffitt, M.C.; Neilan, B.A. Neurotoxic Alkaloids: Saxitoxin and Its Analogs. Mar. Drugs 2010, 8, 2185–2211. [Google Scholar] [CrossRef] [Green Version]
- Yotsu-Yamashita, M.; Kim, Y.H.; Dudley, S.C., Jr.; Choudhary, G.; Pfahnl, A.; Oshima, Y.; Daly, J.W. The structure of zetekitoxin AB, a saxitoxin analog from the Panamanian golden frog Atelopus zeteki: A potent sodium-channel blocker. Proc. Natl. Acad. Sci. USA 2004, 101, 4346–4351. [Google Scholar] [CrossRef] [Green Version]
- Fuhrman, F.A.; Fuhrman, G.J.; Mosher, H.S. Toxin from Skin of Frogs of the Genus Atelopus: Differentiation from Dendrobatid Toxins. Science 1969, 165, 1376–1377. [Google Scholar] [CrossRef]
- Shindelman, J.; Mosher, H.S.; Fuhrman, F.A. Atelopidtoxin from the Panamanian frog, Atelopus Zeteki. Toxicon 1969, 7, 315–319. [Google Scholar] [CrossRef]
- Tanino, H.; Nakada, T.; Kaneko, Y.; Kishi, Y. A stereospecific total synthesis of dl-saxitoxin. J. Am. Chem. Soc. 1977, 99, 2818–2819. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, P.A.; Martineli, M.J.; Polanc, S. Total synthesis of (±)-saxitoxin. J. Am. Chem. Soc. 1984, 106, 5594–5598. [Google Scholar] [CrossRef]
- Fleming, J.J.; Du Bois, J. A Synthesis of (+)-Saxitoxin. J. Am. Chem. Soc. 2006, 128, 3926–3927. [Google Scholar] [CrossRef]
- Iwamoto, O.; Koshino, H.; Hashizume, D.; Nagasawa, K. Total synthesis of (−)-decarbamoyloxysaxitoxin. Angew. Chem. Int. Ed. 2007, 46, 8625–8628. [Google Scholar] [CrossRef]
- Mulcahy, J.V.; Du Bois, J. A Stereoselective Synthesis of (+)-Gonyautoxin 3. J. Am. Chem. Soc. 2008, 130, 12630–12631. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, O.; Shinohara, R.; Nagasawa, K. Total Synthesis of (−)- and (+)-Decarbamoyloxysaxitoxin and (+)-Saxitoxin. Chem. Asian J. 2009, 4, 277–285. [Google Scholar] [CrossRef]
- Iwamoto, O.; Nagasawa, K. Total Synthesis of (+)-Decarbamoylsaxitoxin and (+)-Gonyautoxin 3. Org. Lett. 2010, 12, 2150–2153. [Google Scholar] [CrossRef]
- Sawayama, Y.; Nishikawa, T. A Synthetic Route to the Saxitoxin Skeleton: Synthesis of Decarbamoyl α-Saxitoxinol, an Analogue of Saxitoxin Produc ed by the Cyanobacterium Lyngbya wollei. Angew. Chem. Int. Ed. 2011, 50, 7176–7178. [Google Scholar] [CrossRef]
- Bhonde, V.R.; Looper, R.E. A Stereocontrolled Synthesis of (+)-Saxitoxin. J. Am. Chem. Soc. 2011, 133, 20172–20174. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy, J.V.; Walker, J.R.; Merit, J.E.; Whitehead, A.; Du Bois, J. Synthesis of the Paralytic Shellfish Poisons (+)-Gonyautoxin 2, (+)-Gonyautoxin 3, and (+)-11,11-Dihydroxysaxitoxin. J. Am. Chem. Soc. 2016, 138, 5994–6001. [Google Scholar] [CrossRef] [PubMed]
- Thottumkara, A.P.; Parsons, W.H.; Du Bois, J. Saxitoxin. Angew. Chem. Int. Ed. 2014, 53, 5760–5784. [Google Scholar] [CrossRef] [PubMed]
- Andresen, B.M.; Du Bois, J. De Novo Synthesis of Modified Saxitoxins for Sodium Ion Channel Study. J. Am. Chem. Soc. 2009, 131, 12524–12525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, W.H.; Du Bois, J. Maleimide Conjugates of Saxitoxin as Covalent Inhibitors of Voltage-Gated Sodium Channels. J. Am. Chem. Soc. 2013, 135, 10582–10585. [Google Scholar] [CrossRef]
- Akimoto, T.; Masuda, A.; Yotsu-Mari, M.; Hirokawa, T.; Nagasawa, K. Synthesis of saxitoxin derivatives bearing guanidine and urea groups at C13 and evaluation of their inhibitory activity on voltage-gated sodium channels. Org. Biomol. Chem. 2013, 11, 6642–6649. [Google Scholar] [CrossRef]
- Arakawa, O.; Nishio, S.; Noguchi, T.; Shida, Y.; Onoue, Y. A new saxitoxin analogue from a xanthid crab Atergatis Floridus. Toxicon 1995, 12, 1577–1584. [Google Scholar] [CrossRef]
- Wang, C.; Oki, M.; Nishikawa, T.; Harada, D.; Yotsu-Yamashita, M.; Nagasawa, K. Total Synthesis of 11-Saxitoxinethanoic Acid and Evaluation of its Inhibitory Activity on Voltage-gated Sodium Channels. Angew. Chem. Int. Ed. 2016, 55, 11600–11603. [Google Scholar] [CrossRef]
- Walker, R.J.; Merit, E.J.; Thomas-Tran, R.; Tang, D.T.Y.; Du Bois, J. Divergent Synthesis of Natural Derivatives of (+)-Saxitoxin Including 11-Saxitoxinethanoic Acid. Angew. Chem. Int. Ed. 2018, 58, 1689–1693. [Google Scholar] [CrossRef]
- Paladugu, S.R.; James, C.K.; Looper, R.E. A Direct C11 Alkylation Strategy on the Saxitoxin Core: A Synthesis of (+)-11-Saxitoxinethanoic Acid. Org. Lett. 2019, 21, 7999–8002. [Google Scholar] [CrossRef]
- Lou, J.-Y.; Laezza, F.; Gerber, B.R.; Xiao, M.L.; Yamada, K.A.; Hartmann, H.; Craig, A.M.; Nerbonne, J.M.; Ornitz, D.M. Fibroblast growth factor 14 is an intracellular modulator of voltage-gated sodium channels. J. Physiol. 2005, 569, 179–193. [Google Scholar] [CrossRef]
- Matsuo, J.; Murakami, M. The Mukaiyama Aldol Reaction: 40 Years of Continuous Development. Angew. Chem. Int. Ed. 2013, 52, 9109–9118. [Google Scholar] [CrossRef] [PubMed]
- Mase, N.; Hayashi, Y. The Aldol Reaction: Organocatalysis Approach. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P., Molander, G.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 2, pp. 273–339. [Google Scholar]
- Hosokawa, S. Recent development of vinylogous Mukaiyama aldol reactions. Tetrahedron Lett. 2018, 59, 77–88. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Narasaka, K.; Banno, K. New aldol type reaction. Chem. Lett. 1973, 1011–1014. [Google Scholar]
- Mukaiyama, T.; Banno, K.; Narasaka, K. New cross-aldol reactions. Reactions of silyl enol ethers with carbonyl compounds activated by titanium tetrachloride. J. Am. Chem. Soc. 1974, 96, 7503–7509. [Google Scholar] [CrossRef]
- Noyori, R.; Yokoyama, K.; Sakata, J.; Kuwajima, I.; Nakamura, E.; Shimizu, M. Fluoride ion catalyzed aldol reaction between enol silyl ethers and carbonyl compounds. J. Am. Chem. Soc. 1977, 99, 1265–1267. [Google Scholar] [CrossRef]
- Gingras, M.; Chabre, Y.M.; Raimundo, J.-M. Tetrabutylammonium Difluorotriphenylstannate [Bu4N] [Ph3SnF2]: Delivering Carbon or Fluorine Ligands via Hypercoordination. Synthesis 2006, 1, 182–185. [Google Scholar] [CrossRef]
- Hama, T.; Liu, X.; Culkin, D.A.; Hartwig, J.F. Palladium-Catalyzed α-Arylation of Esters and Amides under More Neutral Conditions. J. Am. Chem. Soc. 2003, 125, 11176–11177. [Google Scholar] [CrossRef]
- Moradi, W.A.; Buchwald, S.L. Palladium-Catalyzed α-Arylation of Esters. J. Am. Chem. Soc. 2001, 123, 7996–8002. [Google Scholar] [CrossRef]
- Magauer, T.; Mulzer, J.; Tiefenbacher, K. Total Syntheses of (+)-Echinopine A and B: Determination of Absolute Stereochemistry. Org. Lett. 2009, 11, 5306–5309. [Google Scholar] [CrossRef]
- Xiao, Q.; Ren, W.-W.; Chen, Z.-X.; Sun, T.-W.; Li, Y.; Ye, Q.-D.; Gong, J.-X.; Meng, F.-K.; You, L.; Liu, Y.-F.; et al. Diastereoselective Total Synthesis of (±)-Schindilactone A. Angew. Chem. Int. Ed. 2011, 50, 7373–7377. [Google Scholar] [CrossRef]
- Negishi, E. Novel and selective α-substitution of ketones and other carbonyl compounds based on Pd-catalyzed cross coupling of α-unsaturated carbonyl derivatives containing α-halogen or α-metal groups. J. Organomet. Chem. 1999, 576, 179–194. [Google Scholar] [CrossRef]
- Johnson, C.R.; Adams, J.P.; Braun, M.P.; Senanayake, C.B.W. Modified stille coupling utilizing α-iodoenones. Tetrahedron Lett. 1992, 33, 919–922. [Google Scholar] [CrossRef]
- Cordvilla, C.; Bartolome, C.; Martinez-Ilarduya, J.M.; Espinet, P. The Stille Reaction, 38 Years Later. ACS Catal. 2015, 5, 3040–3053. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Stoltz, B.; Corey, E.J. Cuprous Chloride Accelerated Stille Reactions. A General and Effective Coupling System for Sterically Congested Substrates and for Enantioselective Synthesis. J. Am. Chem. Soc. 1999, 121, 7600–7605. [Google Scholar] [CrossRef]
- Devlin, A.S.; Du Bois, J. Modular synthesis of the pentacyclic core of batrachotoxin and select batrachotoxin analogue designs. Chem. Sci. 2013, 4, 1059–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allred, G.D.; Liebeskind, L.S. Copper-Mediated Cross-Coupling of Organostannanes with Organic Iodides at or below Room Temperature. J. Am. Chem. Soc. 1996, 118, 2748–2749. [Google Scholar] [CrossRef]
- Colomer, I.; Velado, M.; Fe rnandez de la Pradilla, R.; Viso, A. From Allylic Sulfoxides to Allylic Sulfenates: Fifty Years of a Never-Ending [2,3]-Sigmatropic Rearrangement. Chem. Rev. 2017, 117, 14201–14243. [Google Scholar] [CrossRef]
- Rojas, C.M. The Mislow-Evans rearrangement. In Molecular Rearrangements in Organic Synthesis; Rojas, C.M., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar] [CrossRef]
- Johnson, C.R.; Adams, J.P.; Braun, M.P.; Senanayake, C.B.W.; Wovkulich, P.M.; Uskokovic, M.R. Direct α-iodination of cycloalkenones. Tetrahedron Lett. 1992, 33, 917–918. [Google Scholar] [CrossRef]
- Djuardi, E.; Bovonsombat, P.; McNelis, E. Formations of α-Iodoenones by Iodine and Catalytic Amounts of Amines. Synth. Commun. 1997, 27, 2497–2503. [Google Scholar] [CrossRef]
- Adachi, K.; Yamada, T.; Ishizuka, H.; Oki, M.; Tsunogae, S.; Shimada, N.; Chiba, O.; Orihara, T.; Hidaka, M.; Hirokawa, T.; et al. Synthesis of C12-keto saxitoxin derivatives with unusual inhibitory activity against voltage-gated sodium channels. Chem. Eur. J. 2019. [Google Scholar] [CrossRef]
- Nishikawa, T.; Urabe, D.; Isobe, M. Syntheses of N-Acylisoxazolidine Derivatives, Related to a Partial Structure Found in Zetekitoxin AB, a Golden Frog Poison. Heterocycles 2009, 79, 379–385. [Google Scholar] [CrossRef]
- Paladugu, S.R.; Looper, R.E. Preparation of a 1,2-isoxazolidine synthon for the synthesis of zetekitoxin AB. Tetrahedron Lett. 2015, 56, 6332–6334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernet, B.; Vasella, A. Carbocyclische Verbindungen aus Monosacchariden. II. Umsetzungen in der Mannosereihe. Helv. Chim. Acta 1979, 62, 2400–2410. [Google Scholar] [CrossRef]
- Ferrier, R.J.; Furneaux, R.H.; Prasit, P.; Tyler, P.C. Functionalised carbocycles from carbohydrates. Part 2. The synthesis of 3-oxa-2-azabicyclo[3.3.0]octanes. X-Ray crystal structure of (1R,5S)-6-exo,7-endo,8-exo-triacetoxy-N-methyl-4-endo-phenylthio-3-oxa-2-azabicyclo[3.3.0]octane. J. Chem. Soc. Perkin Trans. 1 1983, 1, 1621–1628. [Google Scholar] [CrossRef]
- Dransfield, P.J.; Moutel, S.; Shipman, M.; Sik, V. Stereocontrolled synthesis of polyhydroxylated hexahydro-1H-cyclopent[c]isoxazoles by intramolecular oxime olefin cycloadditions: An approach to aminocyclopentitols. J. Chem. Soc. Perkin Trans. 1 1999, 1, 3349–3355. [Google Scholar] [CrossRef]
- Ogawa, S.; Orihara, M. Synthesis of the penta-N,O-acetyl derivatives of some pseudo-3-amino-3-deoxy-dl-hexopyranoses a and -dl-hexopyranosylamine derivative. Carbohydr. Res. 1989, 189, 323–330. [Google Scholar] [CrossRef]
- Baumgartner, H.; O’Sullivan, A.C.; Schneider, J. The Synthesis of Oxa-Analogs of the Kainoid Family. Heterocycles 1997, 45, 1537–1549. [Google Scholar]
- Nishikawa, T.; Wang, C.; Akimoto, T.; Koshino, H.; Nagasawa, K. Synthesis of an advance model of zetekitoxin AB focusing on N-acylisoxazolidine amide structure corresponding to C13–C17. Asian J. Org. Chem. 2014, 3, 1308–1311. [Google Scholar] [CrossRef]
- Shibuya, M.; Tomizawa, M.; Suzuki, I.; Iwabuchi, Y. 2-Azaadamantane N-Oxyl (AZADO) and 1-Me-AZADO: Highly Efficient Organocatalysts for Oxidation of Alcohols. J. Am. Chem. Soc. 2006, 128, 8412–8413. [Google Scholar] [CrossRef]
- Shibuya, M.; Sato, T.; Tomizawa, M.; Iwabuchi, Y. Oxoammonium salt/NaClO2: An expedient, catalytic system for one-pot oxidation of primary alcohols to carboxylic acids with broad substrate applicability. Chem. Commun. 2009, 13, 1739–1741. [Google Scholar] [CrossRef]
- Kunishima, M.; Kawachi, C.; Hioki, K.; Tani, S. Formation of carboxamides by direct condensation of carboxylic acids and amines in alcohols using a new alcohol- and water-soluble condensing agent: DMT-MM. Tetrahedron 2001, 57, 1551–1558. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NaV Isoform | Primary Locations | Related Diseases | TTX IC50 (nM) |
|---|---|---|---|
| TTX-sensitive | |||
| NaV1.1 | CNS, PNS, heart | Epilepsy | 5.9 |
| NaV1.2 | CNS | Epilepsy | 7.8 |
| NaV1.3 | Embryonic CNS, injured DRG | Nerve injury | 2.0 |
| NaV1.4 | Skeletal muscle | Myotonia | 4.5 |
| NaV1.6 | CNS, PNS, SMCs, DRG | CNS disorders | 3.8 |
| NaV1.7 | PNS, DRG | Pain sensation | 5.5 |
| TTX-resistant | |||
| NaV1.5 | Heart, embryonic CNS | Cardiac arrhythmias | 1970 |
| NaV1.8 | PNS, DRG | Pain sensation | 1330 |
| NaV1.9 | PNS, DRG | Pain sensation | 59,600 |

| Entry. | SM | R2 | 12 (E:Z) a | Yield (%) |
|---|---|---|---|---|
| 1 | 11a | CO2Et | 12a (5:1) | 96 |
| 2 | 11b | CO2Et | 12b (5:1) | 85 |
| 3 | 11a | 4-MeC6H4 | 12c (> 1:1) | 45 |
| 4 | 11a | 3-FC6H4 | 12d (> 10:1) | 63 |
| 5 | 11a | 4-ClC6H4 | 12e (> 10:1) | 65 |
| 6 | 11a | 4-NO2C6H4 | 12f (6:1) | 80 |
| 7 | 11a | 2-Furyl | 12g (> 10:1) | 80 |
| 8 | 11a | C6H5 | 12h (> 10:1) | 60 |
| 9 | 11b | C6H5 | 12i (E:Z) a | 42 |

| Entry | Conditions | R | Result |
|---|---|---|---|
| 1 | CH2C(OZnBr)OtBu, Pd2(dba)3/dppf, THF | CH2CO2tBu | 18a (decomp.) |
| 2 | CH2C(OSnnBu3)OEt, PdCl2(P(o-tol)3)2, CuF2 | CH2CO2Et | 18b (N.R.) |
| 3 | nBu3SnCH=CH2, Pd(PPh3)4, CuI | CH=CH2 | 18c (< 5%) |
| 4 | nBu3SnCH=CH(OEt), Pd(PPh3)4, CuCl, LiCl, THF | CH=CH(OEt) | 18d (67%) |
| 5 | nBu3SnCH=C(OEt)2, Pd(PPh3)4, CuCl, LiCl, THF | CH=C(OEt)2 | 19 (0–40%) |
| 6 | nBu3SnCH=C(OEt)2, Pd(PPh3)4, CuTC, THF | CH=C(OEt)2 | 19 (60%) |
| Compound | IC50 (mean ± SD) (nM) | n |
|---|---|---|
| dcSTX (3) | 89 ± 36 | 3 |
| SEA (9) | 47 ± 12 | 3 |
| dcSEA (31) | 5700 ± 3.1 | 3 |
| SEE (32) | 185 ± 74 | 4 |
| 11-benzylidene STX (33a) | 16 ± 6.9 | 5 |
| Compound | hNaV1.2 | hNaV1.5 | hNaV1.7 |
|---|---|---|---|
| 11-benzylidene STX (33a) | 5.2 ± 6.0 | 94.1 ± 12.0 | 124.1 ± 20.6 |
| 11-methylbenzylidene STX (33b) | 22.9 ± 8.6 | >300 | >300 |
| 11-fluorobenzylidene STX (33c) | 7.7 ± 1.6 | >300 | >300 |
| 11-nitrobenzylidene STX (33d) | 8.79 ± 0.96 | 50.9 ± 7.8 | >300 |
| 11-furfuryl STX (33e) | 542.7 ± 65.7 | >300 | >300 |
| 11-metoxybenzylidene STX (33f) | 45.0 ±2.72 | >300 | >300 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adachi, K.; Ishizuka, H.; Odagi, M.; Nagasawa, K. Synthetic Approaches to Zetekitoxin AB, a Potent Voltage-Gated Sodium Channel Inhibitor. Mar. Drugs 2020, 18, 24. https://doi.org/10.3390/md18010024
Adachi K, Ishizuka H, Odagi M, Nagasawa K. Synthetic Approaches to Zetekitoxin AB, a Potent Voltage-Gated Sodium Channel Inhibitor. Marine Drugs. 2020; 18(1):24. https://doi.org/10.3390/md18010024
Chicago/Turabian StyleAdachi, Kanna, Hayate Ishizuka, Minami Odagi, and Kazuo Nagasawa. 2020. "Synthetic Approaches to Zetekitoxin AB, a Potent Voltage-Gated Sodium Channel Inhibitor" Marine Drugs 18, no. 1: 24. https://doi.org/10.3390/md18010024