Transcriptomic Analysis of Marine Gastropod Hemifusus tuba Provides Novel Insights into Conotoxin Genes

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. High-Throughput Sequencing and De Novo Assembly
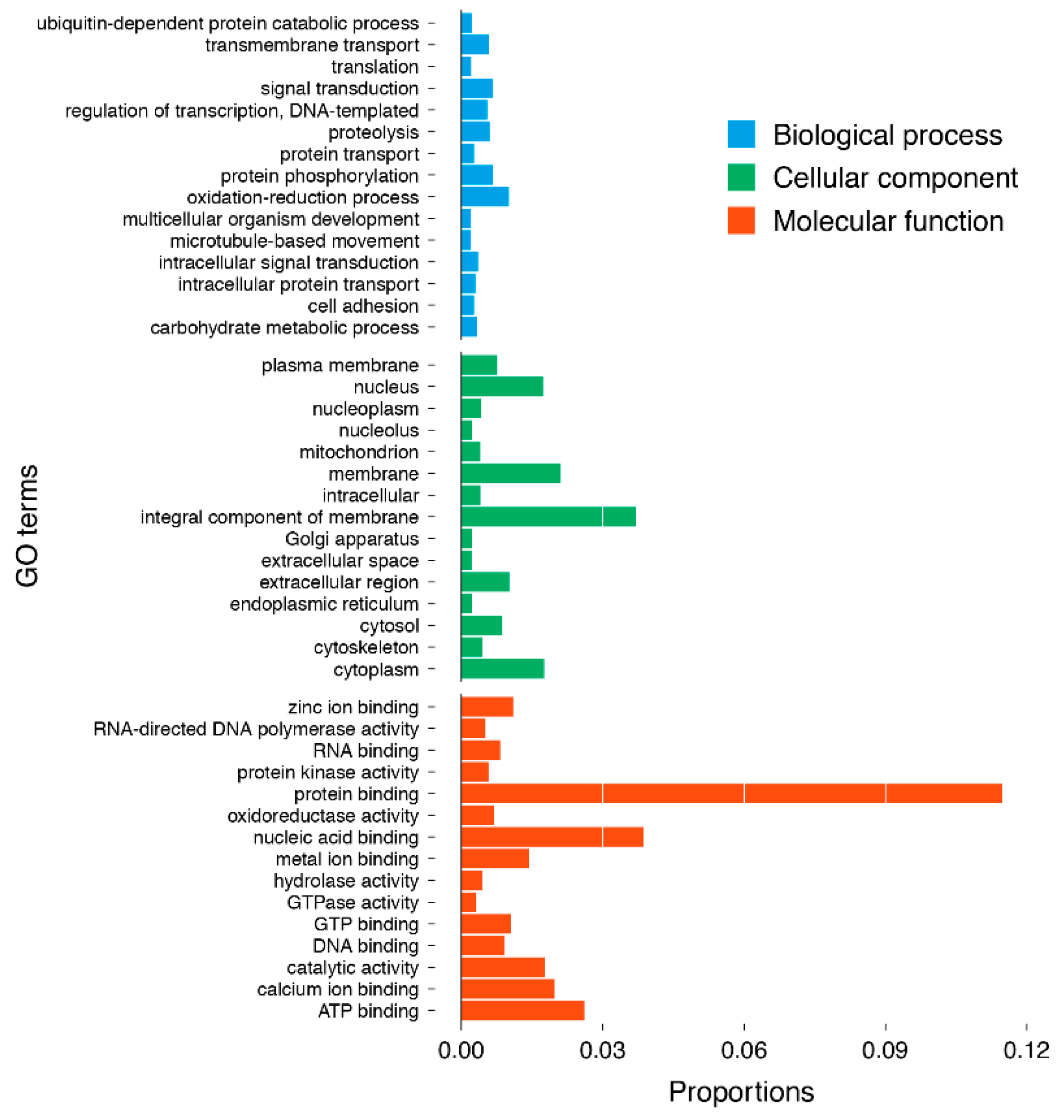
2.2. Annotation and Functional Characterisation of the H. tuba Transcriptome
2.3. Conopeptide Identification
2.4. Characterisation and Validation of Microsatellites
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. RNA Isolation
4.3. Library Construction and Sequencing
4.4. Quality Control and De Novo Assembly
4.5. Annotation and Functional Classification
4.6. Conotoxin Identification and Classification
4.7. Microsatellite Detection and Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sable, R.; Parajuli, P.; Jois, S. Peptides, Peptidomimetics, and Polypeptides from Marine Sources: A Wealth of Natural Sources for Pharmaceutical Applications. Mar. Drugs 2017, 15, 124. [Google Scholar] [CrossRef] [PubMed]
- Stonik, V.A.; Stonik, I.V. Toxins Produced by Marine Invertebrate and Vertebrate Animals: A Short Review; Gopalakrishnakone, P., Ed.; Springer: Dordrecht, The Netherlands, 2014. [Google Scholar]
- Turner, A.H.; Craik, D.J.; Kaas, Q.; Schroeder, C.I. Bioactive Compounds Isolated from Neglected Predatory Marine Gastropods. Mar. Drugs 2018, 16, 118. [Google Scholar] [CrossRef] [PubMed]
- Depledge, M.; Phillips, D. Circulation, respiration and fluid dynamics in the gastropod mollusc, Hemifusus tuba (Gmelin). J. Exp. Mar. Biol. Ecol. 1986, 95, 1–13. [Google Scholar] [CrossRef]
- Pan, Y.; Mu, C.; Li, Q.; Su, X.; Ou, X.; He, J.; Wu, X.; Chen, D. AFLP analysis revealed differences in genetic diversity of Hemifusus tuba. Period. Ocean Univ. China 2013, 43, 58–62. [Google Scholar]
- Yuan, Y.; Li, Q.; Pan, Y.; Kong, L.-F. Isolation and characterization of microsatellite loci in the tuba false fusus (Hemifusus tuba Gmelin). Conserv. Genet. Resour. 2009, 1, 9–11. [Google Scholar] [CrossRef]
- Wu, L.; Li, R.; Wang, C.; Mu, C.; Song, W. Isolation and characterization of 42 microsatellite loci from the Hemifusus tuba Gmelin. Conserv. Genet. Resour. 2014, 6, 707–710. [Google Scholar] [CrossRef]
- Ruan, P.; Jiang, X.; Peng, R.; Li, S. Technique probe: Artificial hatching for seawater snail Hemifusus tuba at industrialized scale. J. Ningbo Univ. 2015, 28, 11–15. [Google Scholar]
- Zhou, S.; Jiang, M.; Jiang, X.; Du, X.; Peng, R.; Han, Q. Effects of dietary betaine on feeding, growth performance, tissue nutritional components and digestive enzyme activities of Hemifusus tuba Gmelin. Chin. J. Anim. Nutr. 2018, 30, 3319–3328. [Google Scholar]
- Morton, B. Prey preference, capture and ration in hemifusus tuba (Gmelin) (prosobranchia: Melongenidae). J. Exp. Mar. Biol. Ecol. 1985, 94, 191–210. [Google Scholar] [CrossRef]
- Morton, B. Reproduction, juvenile growth, consumption and the effects of starvation upon the South China Sea whelk Hemifusus tuba (Gmelin) (Prosobranchia: Melongenidae). J. Exp. Mar. Biol. Ecol. 1986, 102, 257–280. [Google Scholar] [CrossRef]
- Mortem, B. Juvenile growth of the South China Sea whelk Hemifusus tuba (Gmelin) (Prosobranchia: Melongenidae) and the importance of sibling cannibalism in estimates of consumption. J. Exp. Mar. Biol. Ecol. 1987, 109, 1–14. [Google Scholar] [CrossRef]
- Yang, Z.; Li, R.; Mu, C.; Song, W.; Wang, C. Cytological observation of fertilization and analyse of the nurse eggs in Hemifusus tuba (Gmelin). J. Fish. China 2016, 40, 1674–1682. [Google Scholar] [CrossRef]
- Liang, Y.; Zhao, J.; Wu, C. The micro/nanostructure characteristics and the mechanical properties of Hemifusus tuba conch shell. J. Bionic Eng. 2010, 7, 307–313. [Google Scholar] [CrossRef]
- Kaas, Q.; Westermann, J.-C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using ConoServer. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef] [PubMed]
- Medicinal Fauna of China Group. Medicinal Fauna of China; Tianjin Science and Technology Publishing House: Tianjin, China, 1983. [Google Scholar]
- Patnaik, B.B.; Wang, T.H.; Kang, S.W.; Hwang, H.-J.; Park, S.Y.; Park, E.B.; Chung, J.M.; Song, D.K.; Kim, C.; Kim, S.; et al. Sequencing, De Novo Assembly, and Annotation of the Transcriptome of the Endangered Freshwater Pearl Bivalve, Cristaria plicata, Provides Novel Insights into Functional Genes and Marker Discovery. PLoS ONE 2016, 11, e0148622. [Google Scholar] [CrossRef]
- De Oliveira, A.L.; Wollesen, T.; Kristof, A.; Scherholz, M.; Redl, E.; Todt, C.; Bleidorn, C.; Wanninger, A. Comparative transcriptomics enlarges the toolkit of known developmental genes in mollusks. BMC Genom. 2016, 17, 905. [Google Scholar] [CrossRef] [PubMed]
- Harney, E.; Dubief, B.; Boudry, P.; Basuyaux, O.; Schilhabel, M.B.; Huchette, S.; Paillard, C.; Nunes, F.L. De novo assembly and annotation of the European abalone Haliotis tuberculata transcriptome. Mar. Genom. 2016, 28, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Verdes, A.; Anand, P.; Gorson, J.; Jannetti, S.; Kelly, P.; Leffler, A.; Simpson, D.; Ramrattan, G.; Holford, M. From Mollusks to Medicine: A Venomics Approach for the Discovery and Characterization of Therapeutics from Terebridae Peptide Toxins. Toxins 2016, 8, 117. [Google Scholar] [CrossRef]
- Yao, G.; Peng, C.; Zhu, Y.; Fan, C.; Jiang, H.; Chen, J.; Cao, Y.; Shi, Q. High-Throughput Identification and Analysis of Novel Conotoxins from Three Vermivorous Cone Snails by Transcriptome Sequencing. Mar. Drugs 2019, 17, 193. [Google Scholar] [CrossRef]
- Park, S.; Patnaik, B.; Kang, S.; Hwang, H.-J.; Chung, J.; Song, D.; Sang, M.; Patnaik, H.; Lee, J.; Noh, M.; et al. Transcriptomic analysis of the endangered Neritid species Clithon retropictus: De novo assembly, functional annotation, and marker discovery. Genes 2016, 7, 35. [Google Scholar] [CrossRef]
- Song, H.; Yu, Z.-L.; Sun, L.-N.; Gao, Y.; Zhang, T.; Wang, H.-Y. De novo transcriptome sequencing and analysis of Rapana venosa from six different developmental stages using Hi-seq 2500. Comp. Biochem. Physiol. Part D Genom. Proteom. 2016, 17, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Mu, H.; Sun, J.; Heras, H.; Chu, K.H.; Qiu, J.-W. An integrated proteomic and transcriptomic analysis of perivitelline fluid proteins in a freshwater gastropod laying aerial eggs. J. Proteom. 2017, 155, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Patnaik, B.B.; Hwang, H.-J.; Park, S.Y.; Chung, J.M.; Song, D.K.; Han, Y.S.; Patnaik, H.H.; Lee, J.B.; Kim, C.; et al. Transcriptome sequencing and de novo characterization of Korean endemic land snail, Koreanohadra kurodana for functional transcripts and SSR markers. Mol. Genet. Genom. 2016, 291, 1999–2014. [Google Scholar] [CrossRef] [PubMed]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.; Lewis, R.J.; Alewood, P.F. Discovery, Synthesis, and Structure–Activity Relationships of Conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; Norton, R.S. Conotoxin Gene Superfamilies. Mar. Drugs 2014, 12, 6058–6101. [Google Scholar] [CrossRef]
- Peng, C.; Yao, G.; Gao, B.-M.; Fan, C.-X.; Bian, C.; Wang, J.; Cao, Y.; Wen, B.; Zhu, Y.; Ruan, Z.; et al. High-throughput identification of novel conotoxins from the Chinese tubular cone snail (Conus betulinus) by multi-transcriptome sequencing. GigaScience 2016, 5, 17. [Google Scholar] [CrossRef]
- Dutertre, S.; Jin, A.-H.; Vetter, I.; Hamilton, B.; Sunagar, K.; Lavergne, V.; Dutertre, V.; Fry, B.G.; Antunes, A.; Venter, D.J.; et al. Evolution of separate predation- and defence-evoked venoms in carnivorous cone snails. Nat. Commun. 2014, 5, 3521. [Google Scholar] [CrossRef] [Green Version]
- Gorson, J.; Ramrattan, G.; Verdes, A.; Wright, E.M.; Kantor, Y.; Srinivasan, R.R.; Musunuri, R.; Packer, D.; Albano, G.; Qiu, W.-G.; et al. Molecular Diversity and Gene Evolution of the Venom Arsenal of Terebridae Predatory Marine Snails. Genome Biol. Evol. 2015, 7, 1761–1778. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, M.B.; de la Rosa, R.A.C.; Falcón, A.; Olivera, B.M.; Heimer de la Cotera, E.P. Peptide pal9a from the venom of the turrid snail Polystira albida from the Gulf of Mexico: Purification, characterization, and comparison with P-conotoxin-like (framework IX) conoidean peptides. Peptides 2009, 30, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Dutertre, S.; Croker, D.; Daly, N.L.; Andersson, Å.; Muttenthaler, M.; Lumsden, N.G.; Craik, D.J.; Alewood, P.F.; Guillon, G.; Lewis, R.J. Conopressin-T fromConus tulipaReveals an Antagonist Switch in Vasopressin-like Peptides. J. Biol. Chem. 2008, 283, 7100–7108. [Google Scholar] [CrossRef]
- Mansbach, R.A.; Travers, T.; McMahon, B.H.; Fair, J.M.; Gnanakaran, S. Snails in Silico: A Review of Computational Studies on the Conopeptides. Mar. Drugs 2019, 17, 145. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, X.; Yu, Z. Analysis of novel immune–related genes and microsatellite markers in the transcriptome of Paphia undulata. J. Oceanol. Limnol. 2019, 37, 1301–1316. [Google Scholar] [CrossRef]
- Cao, S.; Zhu, L.; Nie, H.; Yin, M.; Liu, G.; Yan, X. De novo assembly, gene annotation, and marker development using Illumina paired-end transcriptome sequencing in the Crassadoma gigantea. Gene 2018, 658, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Hou, R.; Bao, Z.; Du, H.; He, Y.; Su, H.; Zhang, Y.; Fu, X.; Jiao, W.; Li, Y.; et al. Transcriptome sequencing of Zhikong scallop (Chlamys farreri) and comparative transcriptomic analysis with Yesso scallop (Patinopecten yessoensis). PLoS ONE 2013, 8, e63927. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.R.; Burke, J.M.; Burke, J. EST-SSRs as a resource for population genetic analyses. Heredity 2007, 99, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- The R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Kaas, Q.; Yu, R.; Jin, A.-H.; Dutertre, S.; Craik, D.J. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef]
- Lavergne, V.; Dutertre, S.; Jin, A.-H.; Lewis, R.J.; Taft, R.J.; Alewood, P.F. Systematic interrogation of the Conus marmoreus venom duct transcriptome with ConoSorter reveals 158 novel conotoxins and 13 new gene superfamilies. BMC Genom. 2013, 14, 708. [Google Scholar] [CrossRef]
- Koua, D.; Brauer, A.; Laht, S.; Kaplinski, L.; Favreau, P.; Remm, M.; Lisacek, F.; Stöcklin, R. ConoDictor: A tool for prediction of conopeptide superfamilies. Nucleic Acids Res. 2012, 40, W238–W241. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F. genepop’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Rice, W.R. Analyzing Tables of Statistical Tests. Evolution 1989, 43, 223–225. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Number/Length |
|---|---|
| Total number of raw PE reads | 33,546,714 |
| Maximum read length (nt) | 90 |
| Pre-process PE reads | 22,892,498 |
| Cleaned PE reads | 21,397,329 |
| Clean bases | 1.9 Gb |
| Transcripts generated (raw) | 329,633 |
| Percentage of read assembled | 82.9% |
| Transcripts (filtered) | 76,306 |
| Percentage of read assembled | 54.5% |
| GC content | 52.9% |
| Maximum transcripts length | 17,498 |
| Minimum transcripts length | 300 |
| Transcripts > 500 bp | 44,171 |
| Transcripts > 1 kb | 17,188 |
| Transcripts > 10 kb | 56 |
| N50 length (bp) | 1014 |
| Mean length (bp) | 824.6 |
| Unigenes | 61,575 |
| N50 length (bp) | 865 * |
| Mean length (bp) | 744.2 * |
| Database | Number annotated |
|---|---|
| PfamA | 60,116 |
| InterPro * | 38,711 |
| SwissProt | 41,468 |
| KEGG | 64,235 |
| GO | 42,819 |
| All | 26,388 |
| Total | 75,620 |
| Cysteine Framework | Conopeptide | Unique Conopeptide |
|---|---|---|
| Unclassified | 9 | 5 |
| NoCys | 2 | 1 |
| I or XXIV | 1 | 1 |
| VIII | 7 | 5 |
| XIV | 3 | 1 |
| XXII | 3 | 1 |
| IX | 48 | 27 |
| SSR Type | SSR Number | Unigenes Number | Occurrence (%) | Total (%) |
|---|---|---|---|---|
| Di-nucleotide | 6957 | 5167 | 11.3 | 33.6 |
| Tri-nucleotide | 11,654 | 8418 | 19.0 | 56.2 |
| Tetra-nucleotide | 1812 | 1358 | 3.0 | 8.7 |
| Penta-nucleotide | 278 | 232 | 0.5 | 1.3 |
| Hexa-nucleotide | 16 | 15 | <0.1% | 0.1 |
| Total | 20,735 | 14,000 | 33.7 | 100.0 |
| Species | Locus | Size Range (bp) | NA | HO | HE |
|---|---|---|---|---|---|
| H. ternatanus | HT4 | 211-219 | 4 | 1.000 | 0.736 |
| HT10 | 209-218 | 4 | 1.000 | 0.690 | |
| HT20 | 179-189 | 6 | 1.000 | 0.762 | |
| HT22 | 138-148 | 6 | 1.000 | 0.782 | |
| HT24 | 212-216 | 3 | 0.250 | 0.232 | |
| HT25 * | 168-180 | 7 | 1.000 | 0.867 | |
| HT27 | 123-137 | 2 | 0.563 | 0.466 | |
| HT28 * | 122-128 | 4 | 1.000 | 0.651 | |
| HT29 | 132-152 | 10 | 1.000 | 0.891 | |
| HT32 | 249-259 | 5 | 0.875 | 0.718 | |
| HT35 | 155-159 | 3 | 0.688 | 0.599 | |
| HT36 * | 249-261 | 6 | 1.000 | 0.835 | |
| HT39 | 141-147 | 4 | 1.000 | 0.736 | |
| R. venosa | HT15 | 126-136 | 6 | 1.000 | 0.794 |
| HT23 | 254-262 | 5 | 0.950 | 0.676 | |
| HT25 * | 168-182 | 8 | 1.000 | 0.876 | |
| HT28 * | 120-124 | 3 | 1.000 | 0.559 | |
| HT31 | 117-125 | 5 | 1.000 | 0.788 | |
| HT36 * | 245-251 | 4 | 1.000 | 0.740 | |
| HT37 | 280-290 | 6 | 1.000 | 0.781 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Bekaert, M.; Wu, L.; Mu, C.; Song, W.; Migaud, H.; Wang, C. Transcriptomic Analysis of Marine Gastropod Hemifusus tuba Provides Novel Insights into Conotoxin Genes. Mar. Drugs 2019, 17, 466. https://doi.org/10.3390/md17080466
Li R, Bekaert M, Wu L, Mu C, Song W, Migaud H, Wang C. Transcriptomic Analysis of Marine Gastropod Hemifusus tuba Provides Novel Insights into Conotoxin Genes. Marine Drugs. 2019; 17(8):466. https://doi.org/10.3390/md17080466
Chicago/Turabian StyleLi, Ronghua, Michaël Bekaert, Luning Wu, Changkao Mu, Weiwei Song, Herve Migaud, and Chunlin Wang. 2019. "Transcriptomic Analysis of Marine Gastropod Hemifusus tuba Provides Novel Insights into Conotoxin Genes" Marine Drugs 17, no. 8: 466. https://doi.org/10.3390/md17080466