Costatone C—A New Halogenated Monoterpene from the New Zealand Red Alga Plocamium angustum

 ,
,  and
and 
Abstract
:1. Introduction
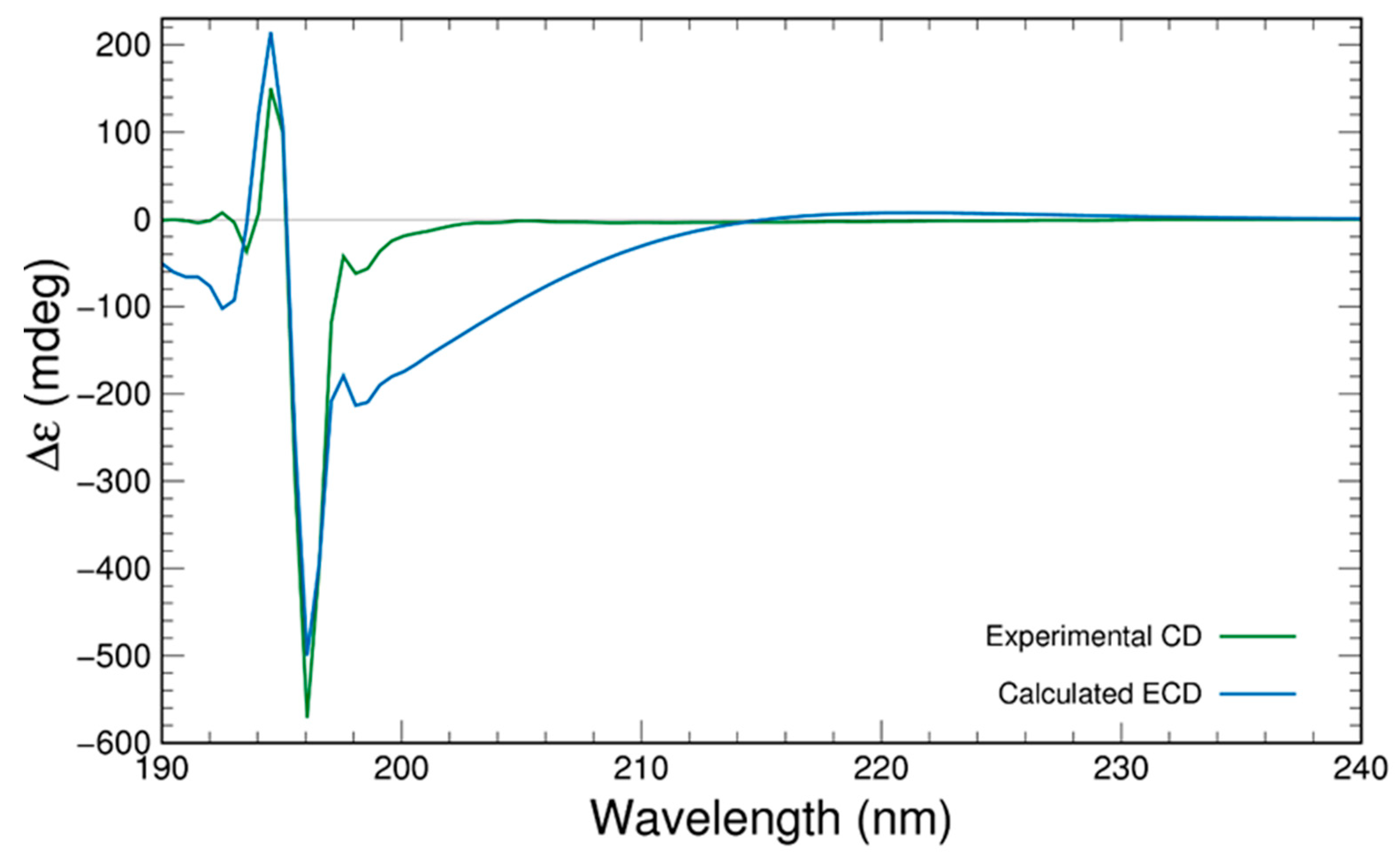
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. General Procedures
4.2. Collection of Plocamium angustum
4.3. Extraction and Isolation
4.4. Preparation of MTPA esters 12a and 12b
4.5. Computational Data
4.6. Antibacterial Bioassay
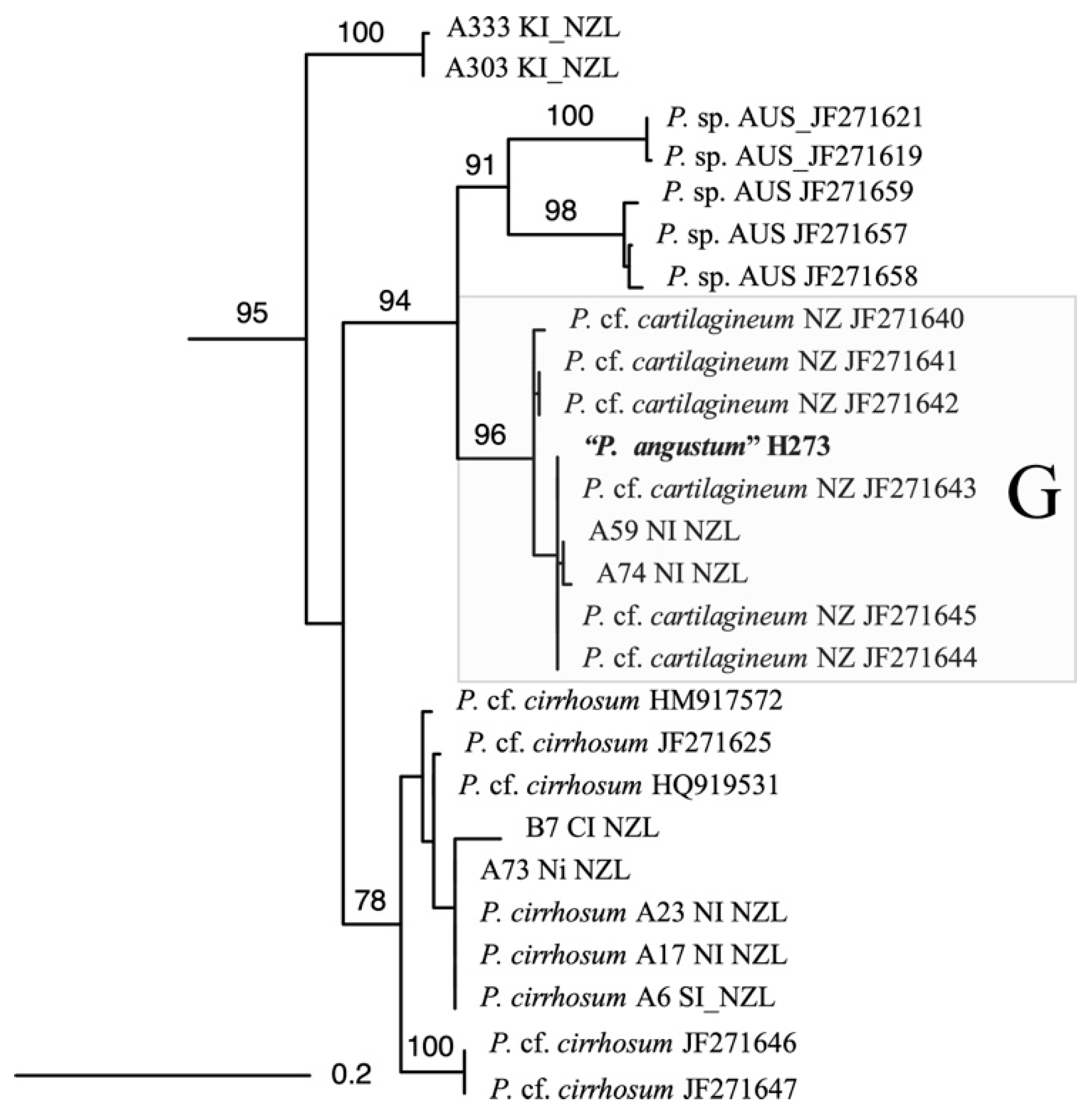
4.7. Molecular Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lefranc, F.; Koutsaviti, A.; Ioannou, E.; Kornienko, A.; Roussis, V.; Kiss, R.; Newman, D. Algae metabolites: From in vitro growth inhibitory effects to promising anticancer activity. Nat. Prod. Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, R.W.; Murphy, P.T.; Wells, R.J. A New Polyhalogenated Monoterpene from the Red Alga Plocamium angustum. Aust. J. Chem. 1979, 32, 2735–2739. [Google Scholar] [CrossRef]
- Timmers, M.A.; Dias, D.A.; Urban, S. Application of HPLC-NMR in the Identification of Plocamenone and Isoplocamenone from the Marine Red Alga Plocamium angustum. Mar. Drugs 2012, 10, 2089–2102. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, R.T.C.; Hall, J.G.; Reiss, J.A. An application of the INEPT pulse sequence to the NMR assignment of halogenated marine natural products. Org. Magn. Reson. 1983, 21, 544–547. [Google Scholar] [CrossRef]
- Cianciola, E.; Popolizio, T.; Schneider, C.; Lane, C. Using Molecular-Assisted Alpha Taxonomy to Better Understand Red Algal Biodiversity in Bermuda. Diversity 2010, 2, 946–958. [Google Scholar] [CrossRef] [Green Version]
- Cremades, J.; Barreiro, R.; Maneiro, I.; Saunders, G.W. A new taxonomic interpretation of the type of Plocamium cartilagineum (Plocamiales, Florideophyceae) and its consequences. Eur. J. Phycol. 2011, 46, 125–142. [Google Scholar] [CrossRef]
- Yano, T.; Kamiya, M.; Arai, S.; Kawai, H. Morphological homoplasy in Japanese Plocamium species (Plocamiales, Rhodophyta) inferred from the Rubisco spacer sequence and intracellular acidity. Phycologia 2004, 43, 383–393. [Google Scholar] [CrossRef]
- Adams, N.M. Seaweeds of New Zealand; Canterbury University Press: Christchurch, New Zealand, 1994; p. 360. [Google Scholar]
- Cooper, M.W. A Taxonomic Investigation into the Red Alga Plocamium within New Zealand. Master’s Thesis, Victoria University of Wellington, Wellington, New Zealand, 2017. [Google Scholar]
- Stierle, D.B.; Wing, R.M.; Sims, J.J. Marine natural products XI costatone and costatolide, new halogenated monoterpenes from the Red seaweed, Plocamium costatum. Tetrahedron Lett. 1976, 17, 4455–4458. [Google Scholar] [CrossRef]
- Motti, C.A.; Thomas-Hall, P.; Hagiwara, K.A.; Simmons, C.J.; Willis, R.; Wright, A.D. Accelerated Identification of Halogenated Monoterpenes from Australian Specimens of the Red Algae Plocamium hamatum and Plocamium costatum. J. Nat. Prod. 2014, 77, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Woolner, V.H.; Jones, C.M.; Field, J.J.; Fadzilah, N.H.; Munkacsi, A.B.; Miller, J.H.; Keyzers, R.A.; Northcote, P.T. Polyhalogenated Indoles from the Red Alga Rhodophyllis membranacea: The First Isolation of Bromo-Chloro-Iodo Secondary Metabolites. J. Nat. Prod. 2016, 79, 463–469. [Google Scholar] [CrossRef]
- Aliev, A.E.; Harris, K.D.M. 37Cl/35Cl isotope effects in 13C NMR spectroscopy of chlorohydrocarbons. Magn. Reson. Chem. 1993, 31, 54–57. [Google Scholar] [CrossRef]
- Bates, P.; Blunt, J.W.; Hartshorn, M.P.; Jones, A.J.; Munro, M.H.G.; Robinson, W.T.; Yorke, S.C. Halogenated Metabolites of the Red Alga Plocamium cruciferum. Aust. J. Chem. 1979, 32, 2545–2554. [Google Scholar] [CrossRef]
- Blunt, J.W.; Bowman, N.J.; Munro, H.G.; Parsons, M.J.; Wright, G.J.; Yeow, K.K. Polyhalogenated Monoterpenes of the New Zealand Marine Red Alga Plocamium cartilagineum. Aust. J. Chem. 1985, 38, 519–525. [Google Scholar] [CrossRef]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the Comparison of Calculated and Experimental Electronic Circular Dichroism Spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Pescitelli, G.; Bruhn, T. Good Computational Practice in the Assignment of Absolute Configurations by TDDFT Calculations of ECD Spectra. Chirality 2016, 28, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polavarapu, P.L.; Covington, C.L. Comparison of Experimental and Calculated Chiroptical Spectra for Chiral Molecular Structure Determination. Chirality 2014, 26, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Dale, J.A.; Dull, D.L.; Mosher, H.S. Alpha-Methoxy-alpha.-trifluoromethylphenylacetic acid, a versatile reagent for the determination of enantiomeric composition of alcohols and amines. J. Org. Chem. 1969, 34, 2543–2549. [Google Scholar] [CrossRef]
- Neises, B.; Steglich, W. Simple Method for the Esterification of Carboxylic Acids. Angew. Chem. 1978, 17, 522–524. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Cammi, R.; Mennucci, B.; Tomasi, J. Fast Evaluation of Geometries and Properties of Excited Molecules in Solution: A Tamm-Dancoff Model with Application to 4-Dimethylaminobenzonitrile. J. Phys. Chem. A 2000, 104, 5631–5637. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V. Solvent effect on vertical electronic transitions by the polarizable continuum model. J. Chem. Phys. 2000, 112, 2427–2435. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V. Time-dependent density functional theory for molecules in liquid solutions. J. Chem. Phys. 2001, 115, 4708–4717. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Polarizable dielectric model of solvation with inclusion of charge penetration effects. J. Chem. Phys. 2001, 114, 5691–5701. [Google Scholar] [CrossRef]
- Cossi, M.; Scalmani, G.; Rega, N.; Barone, V. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 2002, 117, 43–54. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Legault, C.Y. CYLview, 1.0b; Université de Sherbrooke: Sherbrooke, QC, Canada, 2009; Available online: http://www.cylview.org (accessed on 9 April 2019).
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δCa | δHb | COSY | HMBC |
|---|---|---|---|---|
| 1 | 117.5, CH | 6.28 (quin, 1.4) | 3, 9 | 2, 3, 9 |
| 2 | 138.5, C | |||
| 3 | 73.8, CH | 4.22 (dd, 11.7, 2.6) | 1, 4, 9 | 1, 2, 4, 5, 7, 9 |
| 4 | 39.1, CH2 | 2.45 (dt, 12.9, 11.9) | 3, 4, 5 | 2, 3, 5 |
| 2.15 (ddd, 12.9, 4.4, 2.7) | 3, 4, 5 | 3, 5 | ||
| 5 | 54.5, CH | 4.75 (dd, 12.0, 4.4) | 4 | 3, 4, 6, 10 |
| 6 | 73.6, C | |||
| 7 | 83.4, CH | 4.29 (dd, 11.7, 3.9) | 8 | 3, 5, 6, 8, 10 |
| 8 | 29.0, CH2 | 3.94 (t, 11.6) | 7, 8 | 6, 7 |
| 3.73 (dd, 11.6, 3.9) | 7, 8 | 7 | ||
| 9 | 13.1, CH3 | 1.84 (d, 1.4) | 1, 3 | 1, 2, 3 |
| 10 | 28.9, CH3 | 1.70 (s) | 5, 6, 7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bracegirdle, J.; Sohail, Z.; Fairhurst, M.J.; Gerth, M.L.; Zuccarello, G.C.; Ali Hashmi, M.; Keyzers, R.A. Costatone C—A New Halogenated Monoterpene from the New Zealand Red Alga Plocamium angustum. Mar. Drugs 2019, 17, 418. https://doi.org/10.3390/md17070418
Bracegirdle J, Sohail Z, Fairhurst MJ, Gerth ML, Zuccarello GC, Ali Hashmi M, Keyzers RA. Costatone C—A New Halogenated Monoterpene from the New Zealand Red Alga Plocamium angustum. Marine Drugs. 2019; 17(7):418. https://doi.org/10.3390/md17070418
Chicago/Turabian StyleBracegirdle, Joe, Zaineb Sohail, Michael J. Fairhurst, Monica L. Gerth, Giuseppe C. Zuccarello, Muhammad Ali Hashmi, and Robert A. Keyzers. 2019. "Costatone C—A New Halogenated Monoterpene from the New Zealand Red Alga Plocamium angustum" Marine Drugs 17, no. 7: 418. https://doi.org/10.3390/md17070418