Blockade of Human α7 Nicotinic Acetylcholine Receptor by α-Conotoxin ImI Dendrimer: Insight from Computational Simulations


Abstract
:1. Introduction
2. Results and Discussion
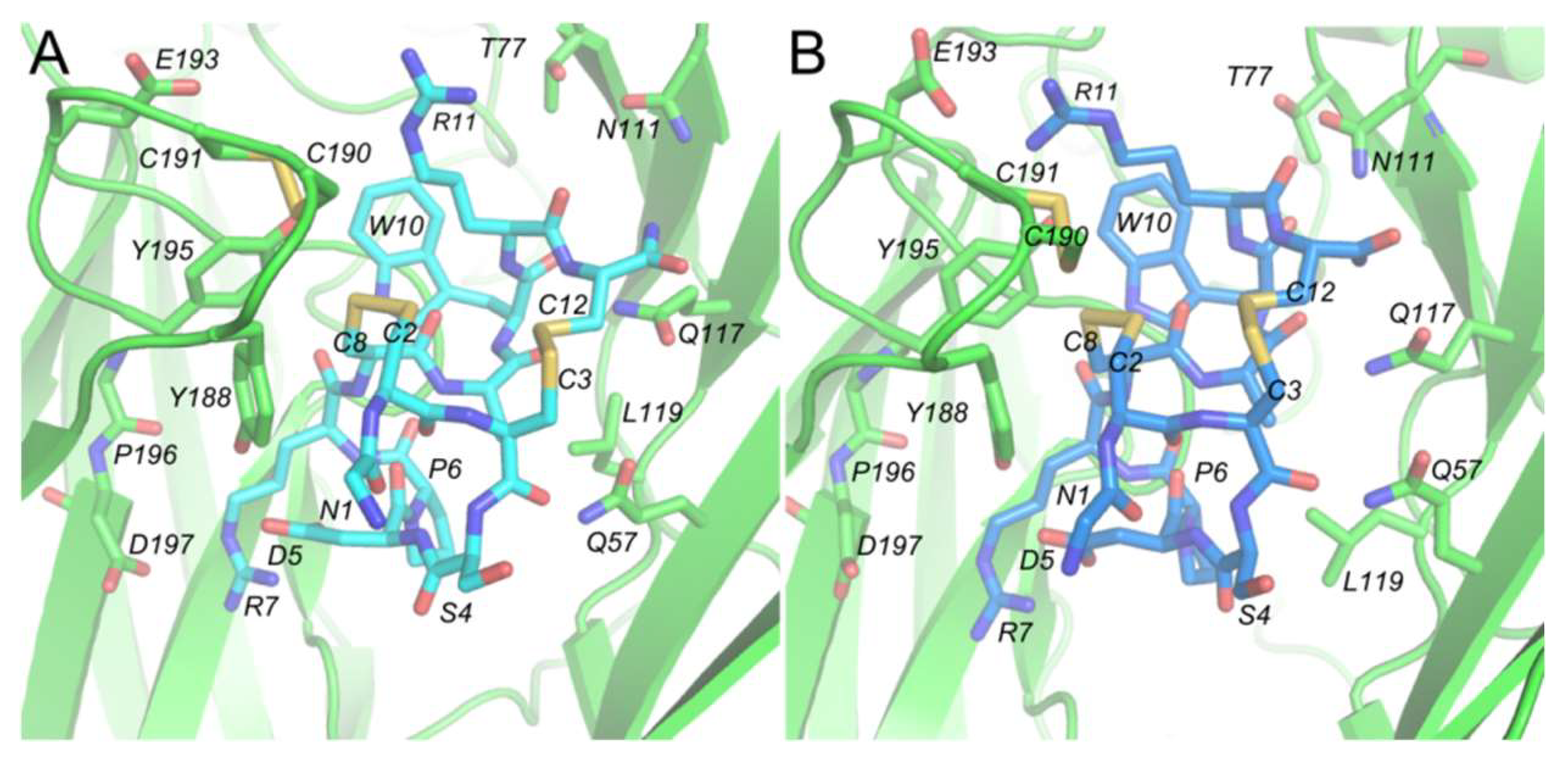
2.1. Comparing the Binding Mode of α-ImI Monomer and α-ImI Dimer at hα7 nAChR
2.2. Effects of the Linker to the Binding of 2×ImI-Dendrimer with hα7 nAChR
2.2.1. Conformation of α-ImI in the 2×ImI-Dendrimer
2.2.2. The Opening of the C-Loop
2.2.3. Interacting with hα7 nAChR
2.3. Binding Energy Calculation and Decomposition
3. Conclusions
4. Materials and Methods
4.1. α-ImI/hα7-nAChR Complex
4.2. 2×ImI-dendrimer/hα7-nAChR Complex
4.2.1. The Building of the 2×ImI-dendrimer
4.2.2. Molecular Dynamics Simulation
4.2.3. Binding Energy Calculations
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hogg, R.C.; Raggenbass, M.; Bertrand, D. Nicotinic acetylcholine receptors: from structure to brain function. Rev. Physiol. Biochem. Pharmacol. 2003, 147, 1–46. [Google Scholar]
- Changeux, J.-P. The nicotinic acetylcholine receptor: the founding father of the pentameric ligand-gated ion channel superfamily. J. Biol. Chem. 2012, 287, 40207–40215. [Google Scholar] [CrossRef]
- Albuquerque, E.X.; Pereira, E.F.R.; Alkondon, M.; Rogers, S.W. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef] [PubMed]
- Isaias, I.U.; Spiegel, J.; Brumberg, J.; Cosgrove, K.P.; Marotta, G.; Oishi, N.; Higuchi, T.; Küsters, S.; Schiller, M.; Dillmann, U.; et al. Nicotinic acetylcholine receptor density in cognitively intact subjects at an early stage of Parkinson’s disease. Front. Aging Neurosci 2014, 6, 213. [Google Scholar] [CrossRef]
- Lombardo, S.; Maskos, U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 2015, 96, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Becchetti, A.; Aracri, P.; Meneghini, S.; Brusco, S.; Amadeo, A. The role of nicotinic acetylcholine receptors in autosomal dominant nocturnal frontal lobe epilepsy. Front. Physiol. 2015, 6, 22. [Google Scholar] [CrossRef] [Green Version]
- Brumwell, C.L.; Johnson, J.L.; Jacob, M.H. Extrasynaptic alpha 7-nicotinic acetylcholine receptor expression in developing neurons is regulated by inputs, targets, and activity. J. Neurosci. 2002, 22, 8101–8109. [Google Scholar] [CrossRef] [PubMed]
- Posadas, I.; López-Hernández, B.; Ceña, V. Nicotinic receptors in neurodegeneration. Curr. Neuropharmacol. 2013, 11, 298–314. [Google Scholar] [CrossRef]
- Taly, A.; Corringer, P.-J.; Guedin, D.; Lestage, P.; Changeux, J.-P. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat. Rev. Drug Discov. 2009, 8, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Westermann, J.-C.; Halai, R.; Wang, C.K.L.; Craik, D.J. ConoServer, a database for conopeptide sequences and structures. Bioinformatics 2008, 24, 445–446. [Google Scholar] [CrossRef]
- Kaas, Q.; Yu, R.; Jin, A.-H.; Dutertre, S.; Craik, D.J. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef]
- Azam, L.; McIntosh, J.M. Alpha-conotoxins as pharmacological probes of nicotinic acetylcholine receptors. Acta Pharmacol. Sin. 2009, 30, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Ellison, M.; Gao, F.; Wang, H.-L.; Sine, S.M.; McIntosh, J.M.; Olivera, B.M. Alpha-conotoxins ImI and ImII target distinct regions of the human alpha7 nicotinic acetylcholine receptor and distinguish human nicotinic receptor subtypes. Biochemistry 2004, 43, 16019–16026. [Google Scholar] [CrossRef]
- Servent, D.; Thanh, H.L.; Antil, S.; Bertrand, D.; Corringer, P.J.; Changeux, J.P.; Ménez, A. Functional determinants by which snake and cone snail toxins block the alpha 7 neuronal nicotinic acetylcholine receptors. J. Physiol. Paris 1998, 92, 107–111. [Google Scholar] [CrossRef]
- Posnett, D.N.; Mcgrath, H.; Tam, J.P. A novel method for producing anti-peptide antibodies. Production of site-specific antibodies to the T cell antigen receptor beta-chain. J. Biol. Chem. 1988, 263, 1719–1725. [Google Scholar]
- Tam, J.P. Synthetic peptide vaccine design: synthesis and properties of a high-density multiple antigenic peptide system. Proc. Nat. Acad. Sci. U. S. A. 1988, 85, 5409–5413. [Google Scholar] [CrossRef]
- Coles, D.J.; Toth, I. Dendritic peptide-based carriers for gene delivery. Curr. Drug Deliv. 2009, 6, 338–342. [Google Scholar] [CrossRef]
- Zhang, C.; Pan, D.; Luo, K.; She, W.; Guo, C.; Yang, Y.; Gu, Z. Peptide dendrimer-Doxorubicin conjugate-based nanoparticles as an enzyme-responsive drug delivery system for cancer therapy. Adv. Healthc. Mater. 2014, 3, 1299–1308. [Google Scholar] [CrossRef]
- De Oliveira, E.; Villén, J.; Giralt, E.; Andreu, D. Synthetic approaches to multivalent lipopeptide dendrimers containing cyclic disulfide epitopes of foot-and-mouth disease virus. Bioconjug. Chem. 2003, 14, 144–152. [Google Scholar] [CrossRef]
- Nardin, E.H.; Calvo-Calle, J.M.; Oliveira, G.A.; Nussenzweig, R.S.; Schneider, M.; Tiercy, J.M.; Loutan, L.; Hochstrasser, D.; Rose, K. A totally synthetic polyoxime malaria vaccine containing Plasmodium falciparum B cell and universal T cell epitopes elicits immune responses in volunteers of diverse HLA types. J. Immunol. 2001, 166, 481–489. [Google Scholar] [CrossRef]
- Schellinger, J.G.; Danan-Leon, L.M.; Hoch, J.A.; Kassa, A.; Srivastava, I.; Davis, D.; Gervay-Hague, J. Synthesis of a trimeric gp120 epitope mimic conjugated to a T-helper peptide to improve antigenicity. J. Am. Chem. Soc. 2011, 133, 3230–3233. [Google Scholar] [CrossRef] [PubMed]
- Skwarczynski, M.; Zaman, M.; Urbani, C.N.; Lin, I.-C.; Jia, Z.; Batzloff, M.R.; Good, M.F.; Monteiro, M.J.; Toth, I. Polyacrylate dendrimer nanoparticles: a self-adjuvanting vaccine delivery system. Angew. Chem. Int. Ed. Engl. 2010, 49, 5742–5745. [Google Scholar] [CrossRef]
- Veprek, P.; Hajdúch, M.; Dzubak, P.; Kuklík, R.; Polakova, J.; Bezouska, K. Comblike dendrimers containing Tn antigen modulate natural killing and induce the production of Tn specific antibodies. J. Med. Chem. 2006, 49, 6400–6407. [Google Scholar] [CrossRef] [PubMed]
- Bay, S.; Cantacuzene, D.; Leclerc, C.; Lo-Man, R. Multiple antigen glycopeptide carbohydrate, vaccine comprising the same and use thereof 2010. US Patent US7696326B2, 3 May 2007. [Google Scholar]
- Tarallo, R.; Carberry, T.P.; Falanga, A.; Vitiello, M.; Galdiero, S.; Galdiero, M.; Weck, M. Dendrimers functionalized with membrane-interacting peptides for viral inhibition. Int. J. Nanomed. 2013, 8, 521–534. [Google Scholar] [Green Version]
- Hatano, K.; Matsubara, T.; Muramatsu, Y.; Ezure, M.; Koyama, T.; Matsuoka, K.; Kuriyama, R.; Kori, H.; Sato, T. Synthesis and influenza virus inhibitory activities of carbosilane dendrimers peripherally functionalized with hemagglutinin-binding Peptide. J. Med. Chem. 2014, 57, 8332–8339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Z.; Xu, X.; Li, Y.; Li, Y.; Jian, Y.; Gu, Z. Bioinspired therapeutic dendrimers as efficient peptide drugs based on supramolecular interactions for tumor inhibition. Angew. Chem. Int. Ed. Engl. 2015, 54, 4289–4294. [Google Scholar] [CrossRef]
- Bonnal, S.; Vigevani, L.; Valcárcel, J. The spliceosome as a target of novel antitumour drugs. Nat. Rev. Drug Discov. 2012, 11, 847–859. [Google Scholar] [CrossRef]
- Henning, L.M.; Bhatia, S.; Bertazzon, M.; Marczynke, M.; Seitz, O.; Volkmer, R.; Haag, R.; Freund, C. Exploring monovalent and multivalent peptides for the inhibition of FBP21-tWW. Beilstein. J. Org. Chem. 2015, 11, 701–706. [Google Scholar] [CrossRef] [Green Version]
- Varamini, P.; Rafiee, A.; Giddam, A.K.; Mansfeld, F.M.; Steyn, F.; Toth, I. Development of New Gonadotropin-Releasing Hormone-Modified Dendrimer Platforms with Direct Antiproliferative and Gonadotropin Releasing Activity. J. Med. Chem. 2017, 60, 8309–8320. [Google Scholar] [CrossRef] [PubMed]
- Yim, C.-B.; Dijkgraaf, I.; Merkx, R.; Versluis, C.; Eek, A.; Mulder, G.E.; Rijkers, D.T.S.; Boerman, O.C.; Liskamp, R.M.J. Synthesis of DOTA-conjugated multimeric [Tyr3]octreotide peptides via a combination of Cu(I)-catalyzed “click” cycloaddition and thio acid/sulfonyl azide “sulfo-click” amidation and their in vivo evaluation. J. Med. Chem. 2010, 53, 3944–3953. [Google Scholar] [CrossRef]
- Livant, D. Compounds for, and methods of, treating cancer and inhibiting invasion and metastases. U.S. Patent 20120077755 A1, 29 March 2012. [Google Scholar]
- Wan, J.; Huang, J.X.; Vetter, I.; Mobli, M.; Lawson, J.; Tae, H.-S.; Abraham, N.; Paul, B.; Cooper, M.A.; Adams, D.J.; et al. α-Conotoxin dendrimers have enhanced potency and selectivity for homomeric nicotinic acetylcholine receptors. J. Am. Chem. Soc. 2015, 137, 3209–3212. [Google Scholar] [CrossRef]
- Jiang, Y.-H.; Emau, P.; Cairns, J.S.; Flanary, L.; Morton, W.R.; McCarthy, T.D.; Tsai, C.-C. SPL7013 gel as a topical microbicide for prevention of vaginal transmission of SHIV89.6P in macaques. AIDS Res. Hum. Retroviruses 2005, 21, 207–213. [Google Scholar] [CrossRef]
- Wan, J.; Mobli, M.; Brust, A.; Muttenthaler, M.; Andersson, Å.; Ragnarsson, L.; Castro, J.; Vetter, I.; Huang, J.X.; Nilsson, M. Synthesis of Multivalent [Lys8]-Oxytocin Dendrimers that Inhibit Visceral Nociceptive Responses. Au. J. Chem. 2017, 70. [Google Scholar] [CrossRef]
- Wan, J.; Brust, A.; Bhola, R.F.; Jha, P.; Mobli, M.; Lewis, R.J.; Christie, M.J.; Alewood, P.F. Inhibition of the norepinephrine transporter by χ-conotoxin dendrimers. J. Pept. Sci. 2016, 22, 280–289. [Google Scholar] [CrossRef]
- Maslennikov, I.V.; Shenkarev, Z.O.; Zhmak, M.N.; Ivanov, V.T.; Methfessel, C.; Tsetlin, V.I.; Arseniev, A.S. NMR spatial structure of α-conotoxin ImI reveals a common scaffold in snail and snake toxins recognizing neuronal nicotinic acetylcholine receptors 1. FEBS Lett. 1999, 444, 275–280. [Google Scholar] [CrossRef]
- Quiram, P.A.; Jones, J.J.; Sine, S.M. Pairwise Interactions between Neuronal α 7 Acetylcholine Receptors and α-Conotoxin ImI. J. Biol. Chem. 1999, 274, 19517–19524. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Craik, D.J.; Kaas, Q. Blockade of neuronal α7-nAChR by α-conotoxin ImI explained by computational scanning and energy calculations. PLoS Comput. Biol. 2011, 7, e1002011. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Wang, H.; Grant, B.; Sine, S.M.; McCammon, J.A. Targeted Molecular Dynamics Study of C-Loop Closure and Channel Gating in Nicotinic Receptors. PLoS Comput. Biol. 2006, 2, e134. [Google Scholar] [CrossRef] [PubMed]
- Brams, M.; Pandya, A.; Kuzmin, D.; van Elk, R.; Krijnen, L.; Yakel, J.L.; Tsetlin, V.; Smit, A.B.; Ulens, C. A Structural and Mutagenic Blueprint for Molecular Recognition of Strychnine and d-Tubocurarine by Different Cys-Loop Receptors. PLoS Biol. 2011, 9, e1001034. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Tae, H.-S.; Wu, G.; Jiang, T.; Yu, R. Exploring the Relationship between Nicotinic Acetylcholine Receptor Ligand Size, Efficiency, Efficacy, and C-Loop Opening. J. Chem. Inf. Model. 2017, 57, 1947–1956. [Google Scholar] [CrossRef] [Green Version]
- Tsui, V.; Case, D.A. Theory and applications of the generalized born solvation model in macromolecular simulations. Biopolymers 2000, 56, 275–291. [Google Scholar] [CrossRef]
- Rastelli, G.; Rio, A.D.; Degliesposti, G.; Sgobba, M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem 2010, 31, 797–810. [Google Scholar] [CrossRef]
- Zhang, X.; Li, X.; Wang, R. Interpretation of the Binding Affinities of PTP1B Inhibitors with the MM-GB/SA Method and the X-Score Scoring Function. J. Chem. Inf. Model. 2009, 49, 1033–1048. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef]
- da Silva Santos, S.; Igne Ferreira, E.; Giarolla, J. Dendrimer Prodrugs. Molecules 2016, 21, 686. [Google Scholar] [CrossRef]
- Le Novere, N.; Grutter, T.; Changeux, J.-P. Models of the extracellular domain of the nicotinic receptors and of agonist- and Ca2+-binding sites. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 3210–3215. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Nistri, A.; Cavalli, A.; Carloni, P. A structural model of agonist binding to the α3β4 neuronal nicotinic receptor. Br. J. Pharmacol. 2003, 140, 921–931. [Google Scholar] [CrossRef]
- Pérez, E.G.; Cassels, B.K.; Zapata-Torres, G. Molecular modeling of the α9α10 nicotinic acetylcholine receptor subtype. Bioorg. Med. Chem. Lett. 2009, 19, 251–254. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinf. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Morris, A.L.; MacArthur, M.W.; Hutchinson, E.G.; Thornton, J.M. Stereochemical quality of protein structure coordinates. Proteins 1992, 12, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Case, D.A.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Greene, D. Amber 2017 Reference Manual; University of California: San Francisco, CA, USA, 2017. [Google Scholar]
- Dupradeau, F.-Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. tools: advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Huang, W.; Jiang, T.; Yu, R. Determination of the μ-Conotoxin PIIIA Specificity Against Voltage-Gated Sodium Channels from Binding Energy Calculations. Mar. Drugs 2018, 16, 153. [Google Scholar] [CrossRef] [PubMed]
- Wittayanarakul, K.; Hannongbua, S.; Feig, M. Accurate prediction of protonation state as a prerequisite for reliable MM-PB(GB)SA binding free energy calculations of HIV-1 protease inhibitors. J. Comput. Chem. 2010, 29, 673–685. [Google Scholar] [CrossRef]
- Dunitz, J.D. Win some, lose some: enthalpy-entropy compensation in weak intermolecular interactions. Chem. Biol. 1995, 2, 709–712. [Google Scholar] [CrossRef] [Green Version]
- Oehme, D.P.; Brownlee, R.T.C.; Wilson, D.J.D. Effect of atomic charge, solvation, entropy, and ligand protonation state on MM-PB(GB)SA binding energies of HIV protease. J. Comput. Chem. 2012, 33, 2566–2580. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands | ΔG (Kcal/mol) | ΔH (Kcal/mol) | TΔS (Kcal/mol) |
|---|---|---|---|
| α-ImI | −30.68 | −73.70 | −43.02 |
| 2×ImI-dendrimer | −78.46 | −233.85 | −155.39 |
| Ligands | ΔG’’’ (Kcal/mol) |
|---|---|
| UNK8 | −22.32 |
| UNK9 | −13.12 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Liang, J.; Zhang, Z.; Jiang, T.; Yu, R. Blockade of Human α7 Nicotinic Acetylcholine Receptor by α-Conotoxin ImI Dendrimer: Insight from Computational Simulations. Mar. Drugs 2019, 17, 303. https://doi.org/10.3390/md17050303
Xu X, Liang J, Zhang Z, Jiang T, Yu R. Blockade of Human α7 Nicotinic Acetylcholine Receptor by α-Conotoxin ImI Dendrimer: Insight from Computational Simulations. Marine Drugs. 2019; 17(5):303. https://doi.org/10.3390/md17050303
Chicago/Turabian StyleXu, Xiaoxiao, Jiazhen Liang, Zheyu Zhang, Tao Jiang, and Rilei Yu. 2019. "Blockade of Human α7 Nicotinic Acetylcholine Receptor by α-Conotoxin ImI Dendrimer: Insight from Computational Simulations" Marine Drugs 17, no. 5: 303. https://doi.org/10.3390/md17050303