Transcriptomic-Proteomic Correlation in the Predation-Evoked Venom of the Cone Snail, Conus imperialis

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Transcriptomic and Bioinformatic Data Analysis
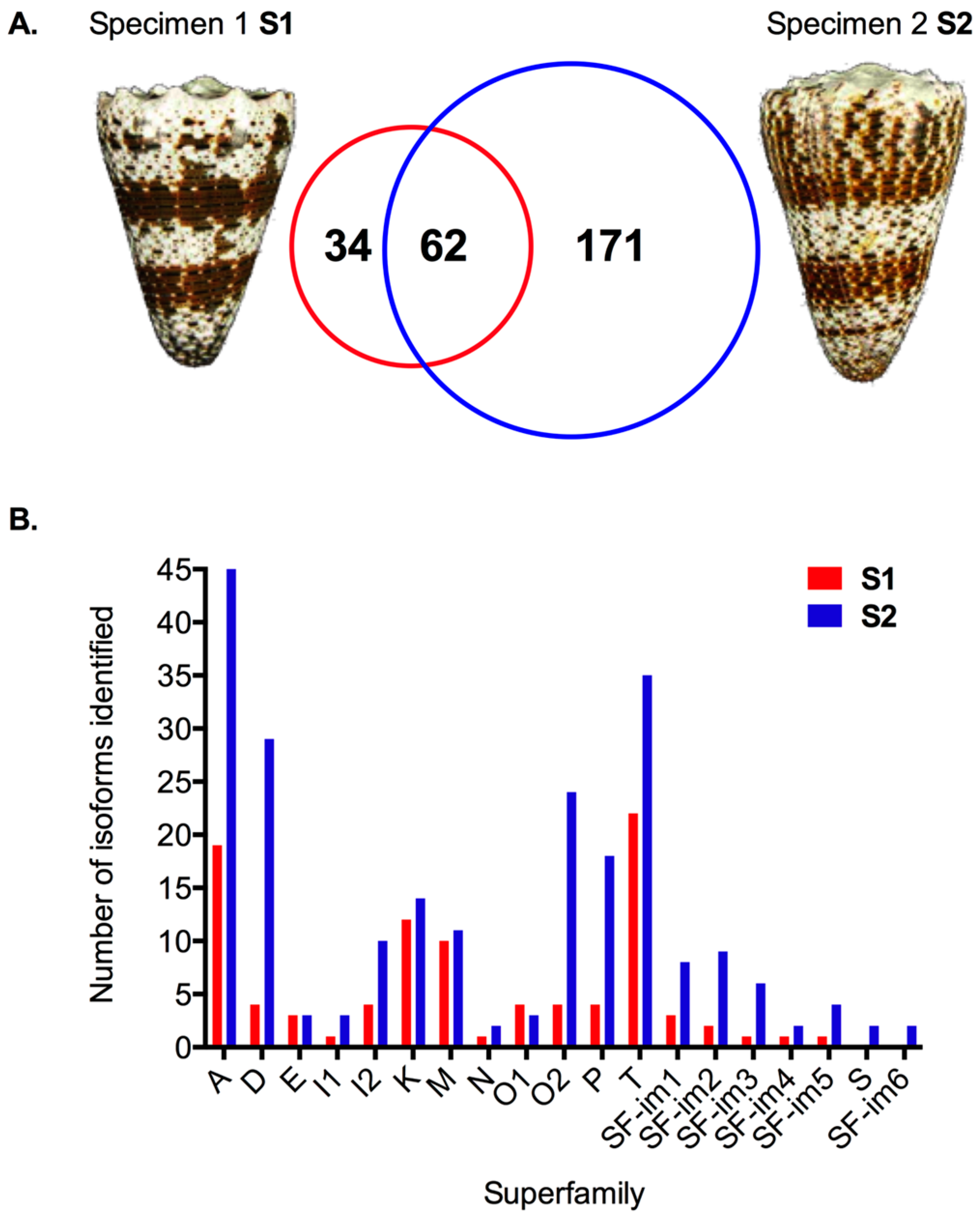
2.2. Transcriptomic Intraspecific Variation
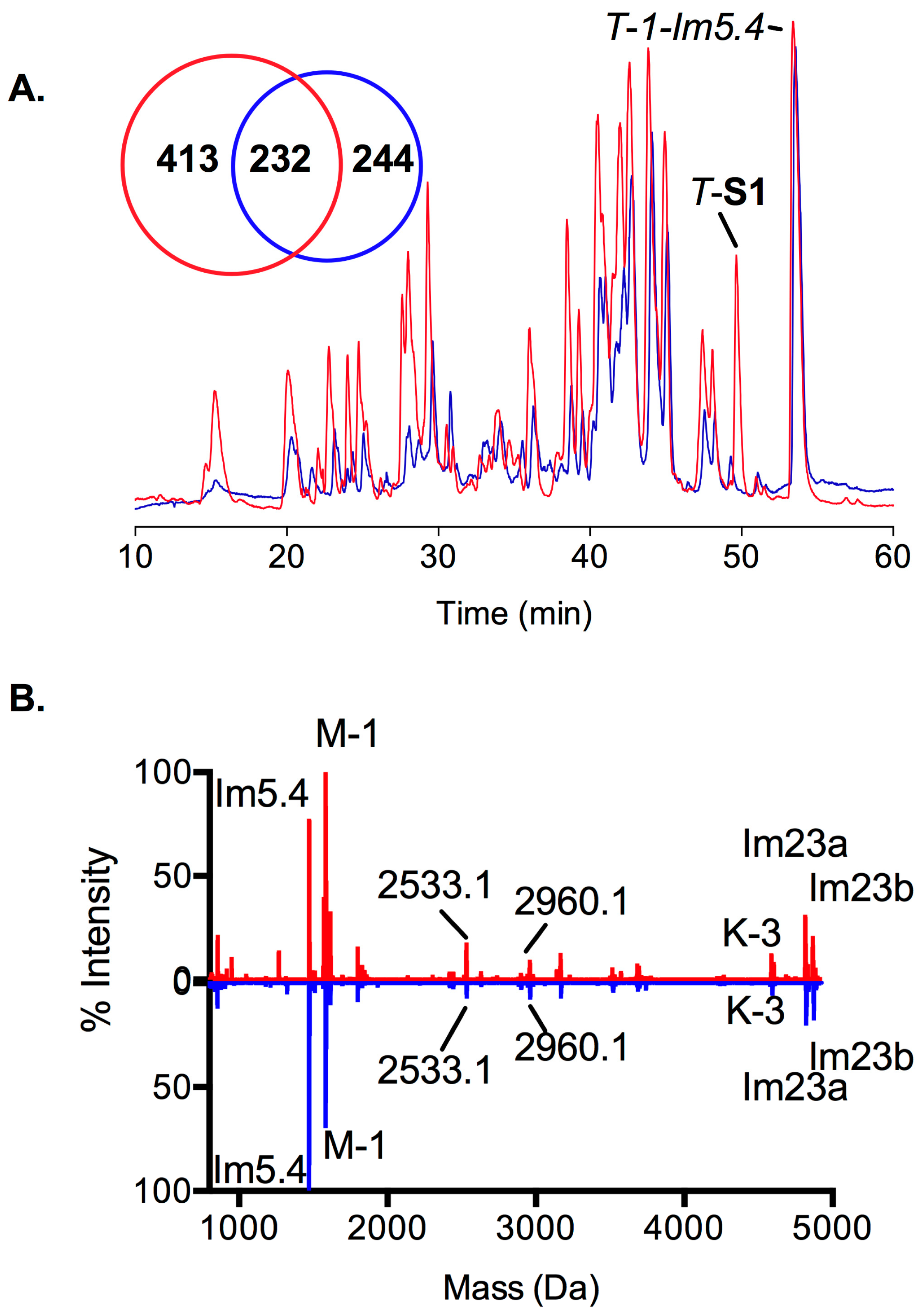
2.3. Proteomic Intraspecific Variation
2.4. Transcription and Translation
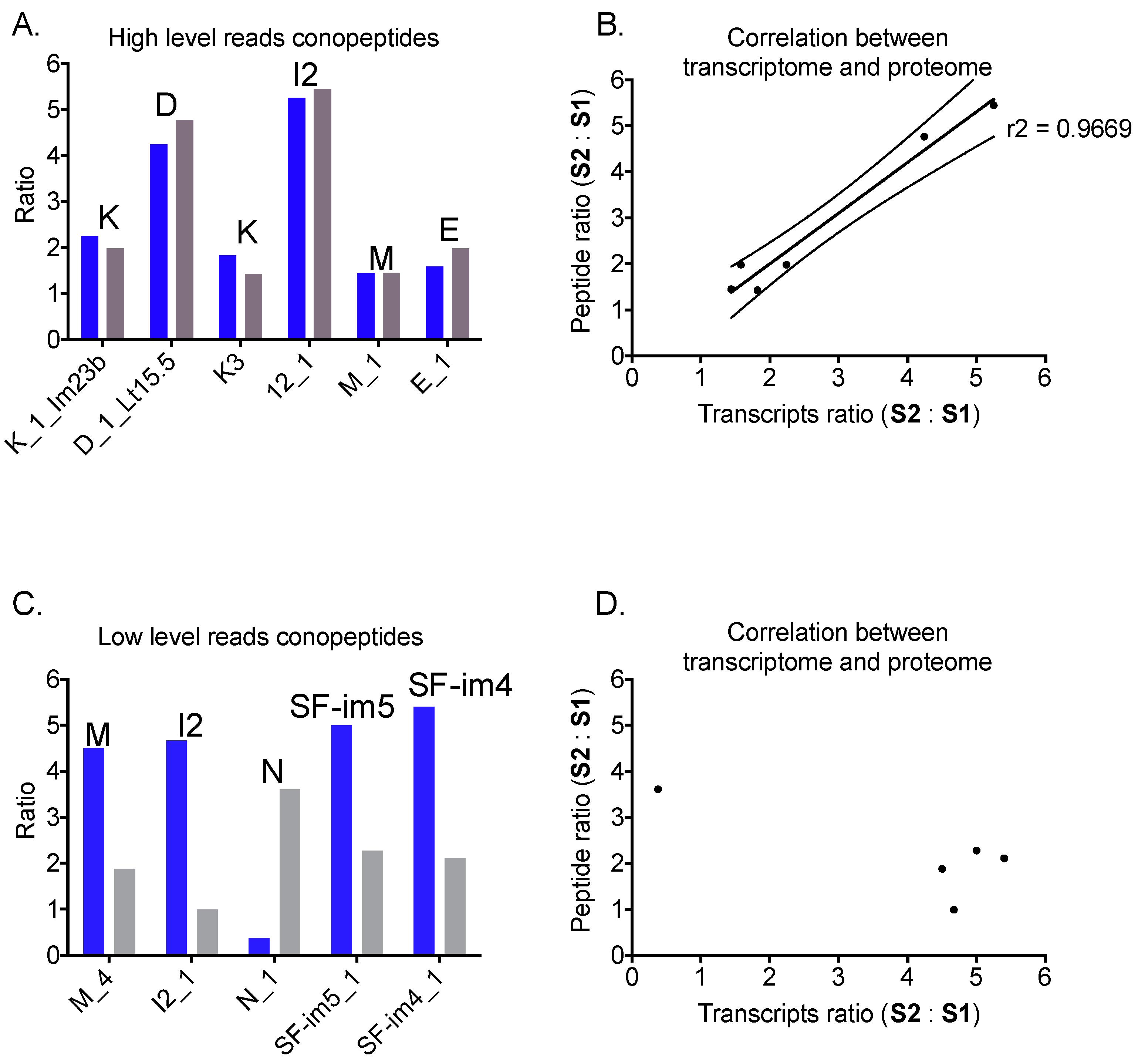
2.5. Correlation between mRNA and Peptide Levels
2.6. Representative Conotoxin Precursors, Mature Peptides, and Cysteine Frameworks by Gene Superfamily
2.6.1. Superfamily A
2.6.2. Superfamily D
2.6.3. Superfamily E
2.6.4. Superfamily I1
2.6.5. Superfamily I2
2.6.6. Superfamily K
2.6.7. Superfamily M
2.6.8. Superfamily N
2.6.9. Superfamily O1
2.6.10. Superfamily O2
2.6.11. Superfamily P
2.6.12. Superfamily S
2.6.13. Superfamily T
2.6.14. Superfamily SF-im1
2.6.15. Superfamily SF-im2
2.6.16. Superfamily SF-im3
2.6.17. Superfamily SF-im4
2.6.18. Superfamily SF-im5
2.6.19. Superfamily SF-im6
3. Discussion
4. Materials and Methods
4.1. Venom Sample Preparation and RNA Extraction
4.2. Proteomic Analysis
4.3. Reduction-Alkylation and Enzyme Digestion
4.4. iTRAQ Labelling, Relative, and Absolute Quantitation of the Injected Venom Samples
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bingham, J.-P.; Jones, A.; Lewis, R.; Andrews, P.; Alewood, P. Conus venom peptides: Inter-species, intra-species and within individual variation revealed by ionspray mass spectrometry. In Biochemical Aspects of Marine Pharmacology; Lazarovici, M.S., Zlotkin, E., Eds.; Alaken Inc.: Anchorage, AK, USA, 1996. [Google Scholar]
- Ruiming, Z.; Yibao, M.; Yawen, H.; Zhiyong, D.; Yingliang, W.; Zhijian, C.; Wenxin, L. Comparative venom gland transcriptome analysis of the scorpion Lychas mucronatus reveals intraspecific toxic gene diversity and new venomous components. BMC Genom. 2010, 11, 452. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, M.A.; Omran, M.A.; Abdel-Nabi, I.M.; Ueda, H.; McVean, A. Intraspecific variation in the Egyptian scorpion Scorpio maurus palmatus venom collected from different biotopes. Toxicon 2009, 53, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Sunagar, K.; Undheim, E.A.; Scheib, H.; Gren, E.C.; Cochran, C.; Person, C.E.; Koludarov, I.; Kelln, W.; Hayes, W.K.; King, G.F.; et al. Intraspecific venom variation in the medically significant southern pacific rattlesnake (Crotalus oreganus helleri): Biodiscovery, clinical and evolutionary implications. J. Proteom. 2014, 99, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Castro, E.N.; Lomonte, B.; del Carmen Gutierrez, M.; Alagon, A.; Gutierrez, J.M. Intraspecies variation in the venom of the rattlesnake Crotalus simus from mexico: Different expression of crotoxin results in highly variable toxicity in the venoms of three subspecies. J. Proteom. 2013, 87, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Lopes, P.H.; Bertani, R.; Goncalves-de-Andrade, R.M.; Nagahama, R.H.; van den Berg, C.W.; Tambourgi, D.V. Venom of the brazilian spider Sicarius ornatus (araneae, sicariidae) contains active sphingomyelinase d: Potential for toxicity after envenomation. PLoS Neglect. Trop. Dis. 2013, 7, e2394. [Google Scholar] [CrossRef] [PubMed]
- Cristina de Oliveira, K.; Goncalves de Andrade, R.M.; Giusti, A.L.; Dias da Silva, W.; Tambourgi, D.V. Sex-linked variation of loxosceles intermedia spider venoms. Toxicon 1999, 37, 217–221. [Google Scholar] [CrossRef]
- Brand, J.M.; Blum, M.S.; Barlin, M.R. Fire ant venoms: Intraspecific and interspecific variation among castes and individuals. Toxicon 1973, 11, 325–331. [Google Scholar] [CrossRef]
- Colinet, D.; Mathe-Hubert, H.; Allemand, R.; Gatti, J.L.; Poirie, M. Variability of venom components in immune suppressive parasitoid wasps: From a phylogenetic to a population approach. J. Insect Physiol. 2013, 59, 205–212. [Google Scholar] [CrossRef]
- Dix, M.W. Snake food preference: Innate intraspecific geographic variation. Science 1968, 159, 1478–1479. [Google Scholar] [CrossRef]
- Gregory-Dwyer, V.M.; Egen, N.B.; Bosisio, A.B.; Righetti, P.G.; Russell, F.E. An isoelectric focusing study of seasonal variation in rattlesnake venom proteins. Toxicon 1986, 24, 995–1000. [Google Scholar] [CrossRef]
- Bordenstein, S.R.; Drapeau, M.D.; Werren, J.H. Intraspecific variation in sexual isolation in the jewel wasp nasonia. Evol. Int. J. Organic Evol. 2000, 54, 567–573. [Google Scholar] [CrossRef]
- Tibbetts, E.A.; Skaldina, O.; Zhao, V.; Toth, A.L.; Skaldin, M.; Beani, L.; Dale, J. Geographic variation in the status signals of Polistes dominulus paper wasps. PLoS ONE 2011, 6, e28173. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; MacDonald, J.C. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Biass, D.; Stocklin, R.; Favreau, P. Dramatic intraspecimen variations within the injected venom of conus consors: An unsuspected contribution to venom diversity. Toxicon 2010, 55, 1453–1462. [Google Scholar] [CrossRef]
- Jakubowski, J.A.; Kelley, W.P.; Sweedler, J.V.; Gilly, W.F.; Schulz, J.R. Intraspecific variation of venom injected by fish-hunting conus snails. J. Exp. Biol. 2005, 208, 2873–2883. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Jin, A.H.; Vetter, I.; Hamilton, B.; Sunagar, K.; Lavergne, V.; Dutertre, V.; Fry, B.G.; Antunes, A.; Venter, D.J.; et al. Evolution of separate predation- and defence-evoked venoms in carnivorous cone snails. Nat. Commun. 2014, 5, 3521. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Jin, A.H.; Dutertre, S.; Kaas, Q.; Lavergne, V.; Kubala, P.; Lewis, R.J.; Alewood, P.F. Transcriptomic messiness in the venom duct of Conus miles contributes to conotoxin diversity. Mol. Cell. Proteom. 2013, 12, 3824–3833. [Google Scholar] [CrossRef]
- Lavergne, V.; Dutertre, S.; Jin, A.H.; Lewis, R.J.; Taft, R.J.; Alewood, P.F. Systematic interrogation of the Conus marmoreus venom duct transcriptome with conosorter reveals 158 novel conotoxins and 13 new gene superfamilies. BMC Genom. 2013, 14, 708. [Google Scholar] [CrossRef]
- Kaas, Q.; Yu, R.; Jin, A.H.; Dutertre, S.; Craik, D.J. Conoserver: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef]
- Allam, A.; Kalnis, P.; Solovyev, V. Karect: Accurate correction of substitution, insertion and deletion errors for next-generation sequencing data. Bioinformatics 2015, 31, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Jin, A.H.; Kaas, Q.; Jones, A.; Alewood, P.F.; Lewis, R.J. Deep venomics reveals the mechanism for expanded peptide diversity in cone snail venom. Mol. Cell. Proteom. 2013, 12, 312–329. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Westermann, J.C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using conoserver. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.M.; Yoshikami, D.; Mahe, E.; Nielsen, D.B.; Rivier, J.E.; Gray, W.R.; Olivera, B.M. A nicotinic acetylcholine receptor ligand of unique specificity, alpha-conotoxin imi. J. Biol. Chem. 1994, 269, 16733–16739. [Google Scholar] [PubMed]
- Nielsen, D.B.; Dykert, J.; Rivier, J.E.; McIntosh, J.M. Isolation of lys-conopressin-g from the venom of the worm-hunting snail, Conus imperialis. Toxicon 1994, 32, 845–848. [Google Scholar] [CrossRef]
- Ellison, M.; McIntosh, J.M.; Olivera, B.M. Alpha-conotoxins ImI and ImII. Similar alpha 7 nicotinic receptor antagonists act at different sites. J. Biol. Chem. 2003, 278, 757–764. [Google Scholar] [CrossRef]
- Hale, J.E.; Butler, J.P.; Gelfanova, V.; You, J.-S.; Knierman, M.D. A simplified procedure for the reduction and alkylation of cysteine residues in proteins prior to proteolytic digestion and mass spectral analysis. Anal. Biochem. 2004, 333, 174–181. [Google Scholar] [CrossRef]
- Loughnan, M.L.; Nicke, A.; Lawrence, N.; Lewis, R.J. Novel alpha d-conopeptides and their precursors identified by cDNA cloning define the d-conotoxin superfamily. Biochemistry 2009, 48, 3717–3729. [Google Scholar] [CrossRef]
- Jimenez, E.C.; Shetty, R.P.; Lirazan, M.; Rivier, J.; Walker, C.; Abogadie, F.C.; Yoshikami, D.; Cruz, L.J.; Olivera, B.M. Novel excitatory conus peptides define a new conotoxin superfamily. J. Neurochem. 2003, 85, 610–621. [Google Scholar] [CrossRef]
- Ye, M.; Khoo, K.K.; Xu, S.; Zhou, M.; Boonyalai, N.; Perugini, M.A.; Shao, X.; Chi, C.; Galea, C.A.; Wang, C.; et al. A helical conotoxin from conus imperialis has a novel cysteine framework and defines a new superfamily. J. Biol. Chem. 2012, 287, 14973–14983. [Google Scholar] [CrossRef]
- Gilly, W.F.; Richmond, T.A.; Duda, T.F., Jr.; Elliger, C.; Lebaric, Z.; Schulz, J.; Bingham, J.P.; Sweedler, J.V. A diverse family of novel peptide toxins from an unusual cone snail, Conus californicus. J. Exp. Biol. 2011, 214, 147–161. [Google Scholar] [CrossRef]
- Dutt, M.; Dutertre, S.; Jin, A.H.; Lavergne, V.; Alewood, P.F.; Lewis, R.J. Venomics reveals venom complexity of the piscivorous cone snail, Conus tulipa. Mar. Drugs 2019, 17, 71. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, H.; Liu, N.; Wu, C.; Jiang, J.; Yue, J.; Jing, Y.; Dai, Q. Diversity and evolution of conotoxins in Conus virgo, Conus eburneus, Conus imperialis and Conus marmoreus from the south china sea. Toxicon 2012, 60, 982–989. [Google Scholar] [CrossRef]
- Biass, D.; Dutertre, S.; Gerbault, A.; Menou, J.L.; Offord, R.; Favreau, P.; Stocklin, R. Comparative proteomic study of the venom of the piscivorous cone snail Conus consors. J. Proteom. 2009, 72, 210–218. [Google Scholar] [CrossRef]
- Dutertre, S.; Jin, A.H.; Alewood, P.F.; Lewis, R.J. Intraspecific variations in Conus geographus defence-evoked venom and estimation of the human lethal dose. Toxicon 2014, 91, 135–144. [Google Scholar] [CrossRef] [PubMed]
- McGivern, J.J.; Wray, K.P.; Margres, M.J.; Couch, M.E.; Mackessy, S.P.; Rokyta, D.R. Rna-seq and high-definition mass spectrometry reveal the complex and divergent venoms of two rear-fanged colubrid snakes. BMC Genom. 2014, 15, 1061. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, C.; Grilley, M.; Miller, C.; Shon, K.J.; Cruz, L.J.; Gray, W.R.; Dykert, J.; Rivier, J.; Yoshikami, D.; Olivera, B.M. A new family of conus peptides targeted to the nicotinic acetylcholine receptor. J. Biol. Chem. 1995, 270, 22361–22367. [Google Scholar] [CrossRef] [PubMed]
- Kohn, A.J.; Hunter, C. The feeding process in Conus imperialis. The Veliger 2011, 44, 232–234. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Bendtsen, J.D.; Nielsen, H.; von Heijne, G.; Brunak, S. Improved prediction of signal peptides: Signalp 3.0. J. Mol. Biol. 2004, 340, 783–795. [Google Scholar] [CrossRef]
- Zieske, L.R. A perspective on the use of itraq reagent technology for protein complex and profiling studies. J. Exp. Bot. 2006, 57, 1501–1508. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene Superfamily 1 | Signal Sequence | Cysteine Patterns | Framework |
|---|---|---|---|
| A | MGMRMMFTVFLLVVLATAVLP | CC-C-C | I |
| D | MPKLEMMLLVLLILPLCYIDA | C-C-C-CC-C-C-C-C-C | Novel |
| E | MMMRVFIAMFFLLALVEA | C-C-C-C-C-C-C-C | XXII |
| I1 | MKLALTFLLILMILPLMTG | C-C-CC-CC-C-C | XI |
| I2 | MFRVTSVLLVIVLLNLVVLTNA | C-C-CC-CC-C-C | XI |
| K | MIMRMTLTLFVLVVMTAASASG | C-C-C-CC-C | XXIII |
| M | MMSTLVVLLTICLLMLPLTA | CC-C-C-CC | III |
| N | MSTLGMMLLILLLLVPLATFA | C-C-CC-CC-C-C-C-C | Novel |
| O1 | MKLRCMMIVAVLFLTASIFITA | C-C-CC-C-C | VI/VII |
| O2 | MKLTILLLVAALLVLTQA | C-C-CC-C-C | VI/VII |
| P | MHLSLASSAALMLLLLFALGNFVGVQP | C-C-C-C-C-C | IX |
| T | MRCLPVVVFLLLLLSAAA | CC-CC | V |
| SF-im1 | MARFLSILLCFAMATGLAAG | C-C | - |
| SF-im2 | MRLTTMHSVILMLLLVFAFDNVDG | C-C-C-C-C-C-CC | Novel |
| SF-im3 | MSKSGMLLFVLLLLLPLAIP | C-CC-C-CC-C-C-C-C | XX |
| SF-im4 | MKFFTCLLLLLVVLTVVFDNVDA | C-C-C-CC-C | XXIII |
| SF-im5 | MKTGMIICLLLIAFMDADG | C-C-CC-C-C | VI/VII |
| S (S2) | MMLKMGAMFAILLLFALSSS | C-C-C-C-C-C-C-C | VIII |
| SF-im6 (S2) | MGVFRCCLAAALVVVCLSRMGG | C-C-C-C | XIV |
| Gene Superfamily | Total Reads | Total Paralogs | Common Paralogs |
|---|---|---|---|
| A | 5826 | 52 | 15 |
| D | 2421 | 29 | 4 |
| E | 204 | 3 | 3 |
| I1 | 55 | 3 | 1 |
| I2 | 1426 | 10 | 4 |
| K | 1032 | 22 | 6 |
| M | 498 | 17 | 4 |
| N | 34 | 2 | 1 |
| O1 | 76 | 6 | 1 |
| O2 | 1038 | 25 | 3 |
| P | 400 | 18 | 4 |
| S | 19 | 2 | 0 |
| T | 2166 | 46 | 11 |
| SF-im1 | 85 | 10 | 1 |
| SF-im2 | 81 | 10 | 1 |
| SF-im3 | 73 | 6 | 1 |
| SF-im4 | 37 | 2 | 1 |
| SF-im5 | 25 | 4 | 1 |
| SF-im6 | 11 | 2 | 0 |
| Name | Mature Peptide Sequence | cDNA Reads | MSMS Precursor Intensity | ||
|---|---|---|---|---|---|
| Full Precursor | Mature Peptide | RM Only | RM and Digested | ||
| T_S1 (ImVC) | FLNTICCWSR1ACCG-NH2 | 41 | 137 | 1839217 | 22,290 |
| T_1_Im5.4 | FLNTICCWSGACCG-NH2 | 491 | 1297 | 7,744,866 | 1,248,633 |
| Ratio | 12 | 9 | 4 | 56 | |
| Name | Seq No. | Precursor Sequence |
|---|---|---|
| A_1 | im001 | MGMRMMFTVFLLVVLATTVVPITLASATDGRNAAADARMSPLISKFKK-DYCHKYGYTIG |
| A_2 | im002 | MGMRMMFTVFLLVVLATTVVPITLASATDGRNAAANARVSPVISKFSKK-WCHPNPYTVG |
| A_3_Bn1.3 | im003 | MGMRMMFTVFLLVVLATAVLPVTLDRASDGRNAAANAKTPRLIAPFIRDYCCHRGPCMVWCG |
| D_1_Lt15.5 | im004 | MPKLEMMLLVLLILPLCYIDAVGPPPPWNMEDEIIEHWQKLHCHEISDLTPWILCSPEPLCGGKGCCAQEVCDCSGPVCTCPPCL |
| E_1 | im005 | MMMRVFIAMFFLLALVEAGWPRLYDKNCKKNILRTYCSNKICGEATKNTNGELQCTMYCRCANGCFRGQYIDWPNQQTNLLFC |
| I1_1 | im006 | MKLALTFLLILMILPLMTGEKTSDDLELRGVESLRAIFRDRRCSDNIGATCSDRFDCCGSMCCIGGQCVVTFAECS |
| I2_1 | im007 | MFRVTSV--LLVIV-LLNLVVLTNACHMD---CSKMT-CCSGICCFY-CGRPMCPGTRRALLQRLVGHQR |
| I2_2 | im008 | MFRLTSVGCILLVIAFLNLVGLTNACTSEGYSCSSDSNCCKNVCCWNVCESH-CRHPGKRTRLQGFFKHRR |
| K_1_Im23b | im009 | MIMRMTLTLFVLVVMTAASASGDALTEAKRIPYCGQTGAECYSWCIKQDLSKDWCCDFVKTIARLPPAHICSQ |
| K_2_Im23a | im010 | MIMRMTLTLFVLVVMTAASASGDALTEAKRIPYCGQTGAECYSWCIKQDLSKDWCCDFVKDIRMNPPADKCP |
| K_3 | im011 | MIMRMTLTLFVLVVMTAASASGDALTEAKRVPYCGQTGAECYSWCKEQHLIR--CCDFVKYVGMNPPADKCR |
| M_1 | im012 | MMSTLVVLLTICLLMLPLTARQLDADQLADQLAERMEDISADQNRWFDPVKRCCMRPICM-----CP-CCVNG |
| M_2_Eu3.3 | im013 | MMSKLGVLLAICLLMLPLTALPLDGDQPQER---KEDGKSAALQPWFDPVKRCCQAA-CSPWL--CLPCCG |
| M_3_Bt3.1 | im014 | MMSTLGVLLTIGLLLFPLTALPLDGDQPADQPAERLQDISPKEIPGSDPFKRCCHAPYCTPPHLGC-PCCGK |
| M_4 | im015 | MSKVGVVPLIFLVLLSIAALQNGDDPRRQRDEKQSPQGDILRSTLTKYSYNIQRRCWAGGSPCHLCSSSQVCIAPTGHPAIMCGRCVPILT |
| N_1 | im016 | MSTLGMMLLILLLLVPLATFADDGPTMRGHRSAKLLAHTTRDSCPSGTNCPSKICCNGNCCSKSSCRCETNQATKERVCVC |
| O1_1_Conotoxin3 | im017 | MKLRCMMIVAVLFLTASIFITADNSRNGIENLPRMRRHEMKKPKASKLNKRGCLPDEYFCGFSMIGALLCCSGWCLGICMT |
| O2_1_im6.2 | im018 | MKLTILLLVAALLVLTQARTERRRVKSRKTSSTYDDEMATFCWSYWNEFQYSYPYTYVQPCLTLGKACTTNSDCCSKYCNTKMCKINWEG |
| O2_3 | im019 | MEKLTILLLVTAVLMSTQALMQSGIEKRQRAKIKFFSKRKTTAER----------WWEGECYDWLRQCSSPAQCCSGNCGAH-CKAW |
| P_1 | im020 | MHLSLASSAALMLLLLFALGNFVGVQPGQIRDLNKGQLKDNRRNLQSQRKQMSLLKSLHDRNG-CNGNTCSNSP-CPNNCY-CDTEDD---CHPDRREH |
| P_2 | im021 | MHLSTASSVALMFFLLFAFYGVQPELMTRDVDNGQLTDNRRNLRSRVKPTGLFKSRK--PSED-C-GKTCETAENCPDDCSSCLSVEGTYRCA |
| P_3 | im022 | MHRSLAGSAVLMLLLLFALGNFVGVQPGLVTRDADNGQLMDNRRNLRLERKTMSLFKSLDKRADCSTY-CFGMGICQSGCY-CGPGHA---CMPNGR |
| T_1_Im5.4 | im023 | MRCLPVVVFLLLLLSAAAAPGVGSKTERLPGLTSSGDSDESLP---FLNTICCWSGA-CCGG |
| T_2 | im024 | MCCIPVFFILLLLIPSAPSILAQPTTKGDVALASSYDDAKRTLQRLSIKYSCCPGIVSCCVIP |
| SF-im1_1 | im025 | MARFLSILLCFAMATGLAAGIRYPDRVLGRCSTHDLSKMEIDTNLDGVYSPHRSFCTCGSGEVYFTAKDRRNHSNYRVYVCGMPTEFCTAENPVRDP |
| SF-im2_1 | im026 | MRLTTMHSVILMLLLVFAFDNVDGDEPGQTARDVDNRNFMSILRSEGKPVHFLRAIKKRDCTGQACTTGDNCPSECVCNEHHFCTGKCCYFLHA |
| SF-im3_1 | im027 | MSKSGMLLFVLLLLLPLAIPELAPAGRSVTHHFRDFGAKRSVPISCVNPSTPNLQGSWQDKKCCSTKLCSPTNCCESSTCSCVEGSCQCL |
| SF-im4_1 | im028 | MKFFTCLLLLLVVLTVVFDNVDACDRSCTGVMGHPSCATCCACFTSAGKRHADGQHSRMKVRTGAKNLLKRMPLH |
| SF-im5_1 | im029 | MKTGMIICLLLIAFMDADGSPGDTLYSQKTADTDSGMKRFQKTFQKRRCVFCPKEPCCDGDQCMTAPGTGPFCG |
| S_1 | im030 | MMLKMGAMFAILLLFALSSSQQEGDVQARKIRLRNDFLRTSRMIFTRGCGGSCHTSPGCGGNCECNSPVPCYCSGTETCVCVCSG |
| SF-im6_1 | im031 | MGVFRCCLAAALVVVCLSRMGGTEPLESNHEDERRADDTSGDDCVDTNEDCVNWASTGQCEANPSYMRENCRK |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, A.-H.; Dutertre, S.; Dutt, M.; Lavergne, V.; Jones, A.; Lewis, R.J.; Alewood, P.F. Transcriptomic-Proteomic Correlation in the Predation-Evoked Venom of the Cone Snail, Conus imperialis. Mar. Drugs 2019, 17, 177. https://doi.org/10.3390/md17030177
Jin A-H, Dutertre S, Dutt M, Lavergne V, Jones A, Lewis RJ, Alewood PF. Transcriptomic-Proteomic Correlation in the Predation-Evoked Venom of the Cone Snail, Conus imperialis. Marine Drugs. 2019; 17(3):177. https://doi.org/10.3390/md17030177
Chicago/Turabian StyleJin, Ai-Hua, Sébastien Dutertre, Mriga Dutt, Vincent Lavergne, Alun Jones, Richard J. Lewis, and Paul F. Alewood. 2019. "Transcriptomic-Proteomic Correlation in the Predation-Evoked Venom of the Cone Snail, Conus imperialis" Marine Drugs 17, no. 3: 177. https://doi.org/10.3390/md17030177