Biochemical and Anti-Triple Negative Metastatic Breast Tumor Cell Properties of Psammaplins

 ,
,
Abstract
:1. Introduction
2. Results
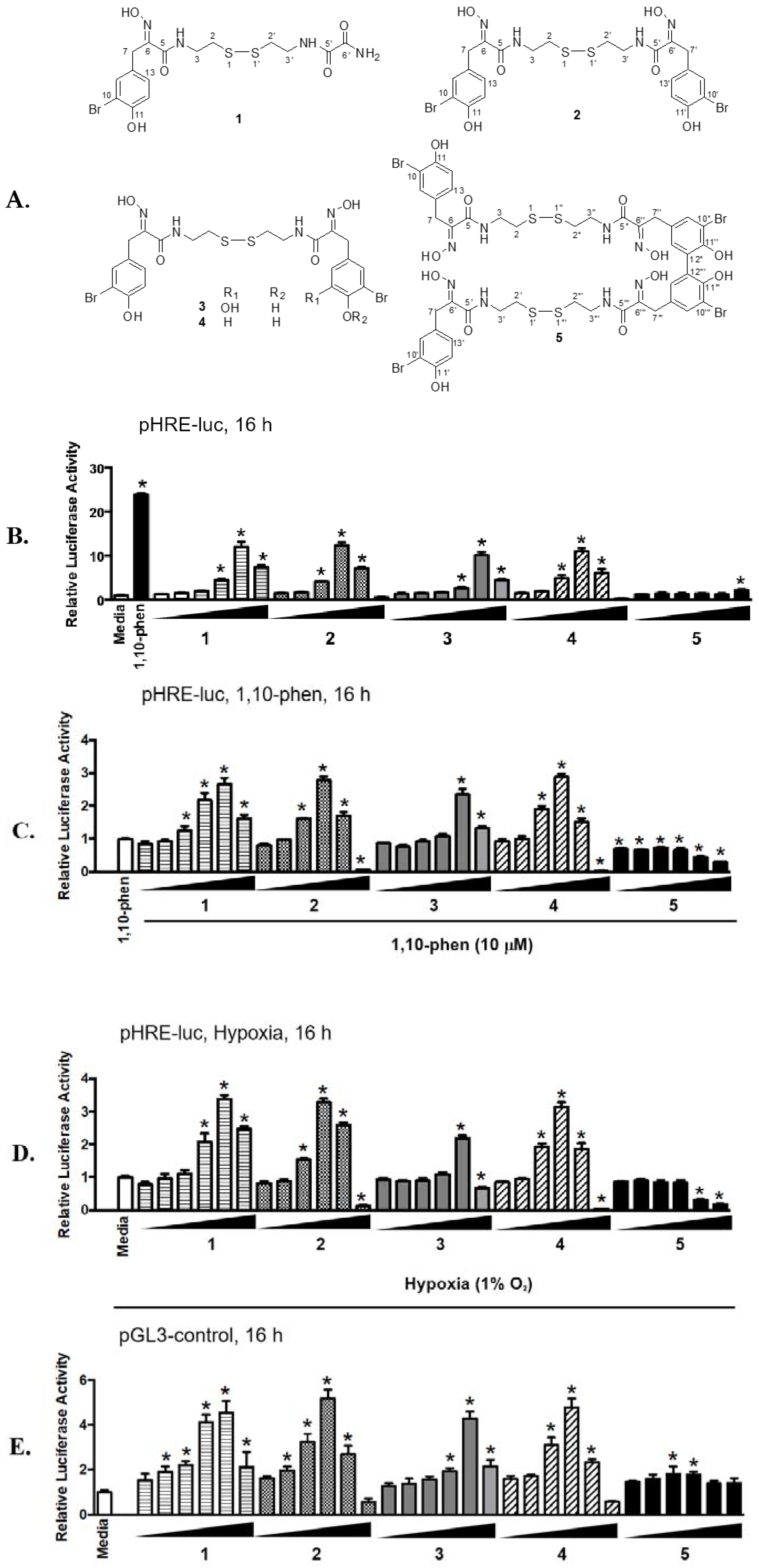
2.1. Psammaplins Exhibit Concentration-Dependent Biphasic Effects on HIF-1 Activity
2.2. Differential HDAC Inhibition by Psammaplin Analogues
2.3. Effects of Psammaplin A on HIF-1 Target Gene Expression
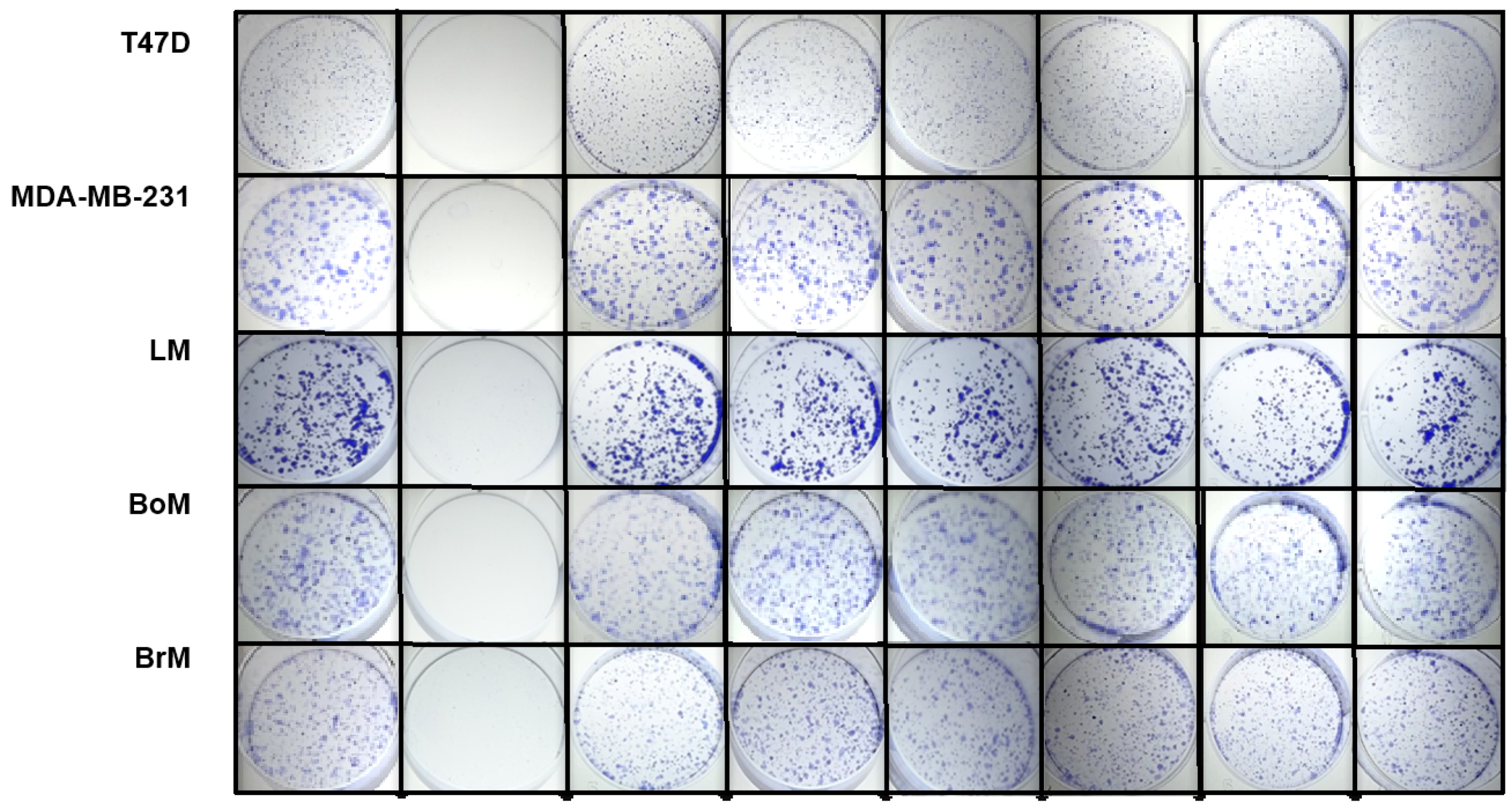
2.4. Psammaplins Suppress Cell Proliferation/Viability in a Cell Line-Dependent Manner
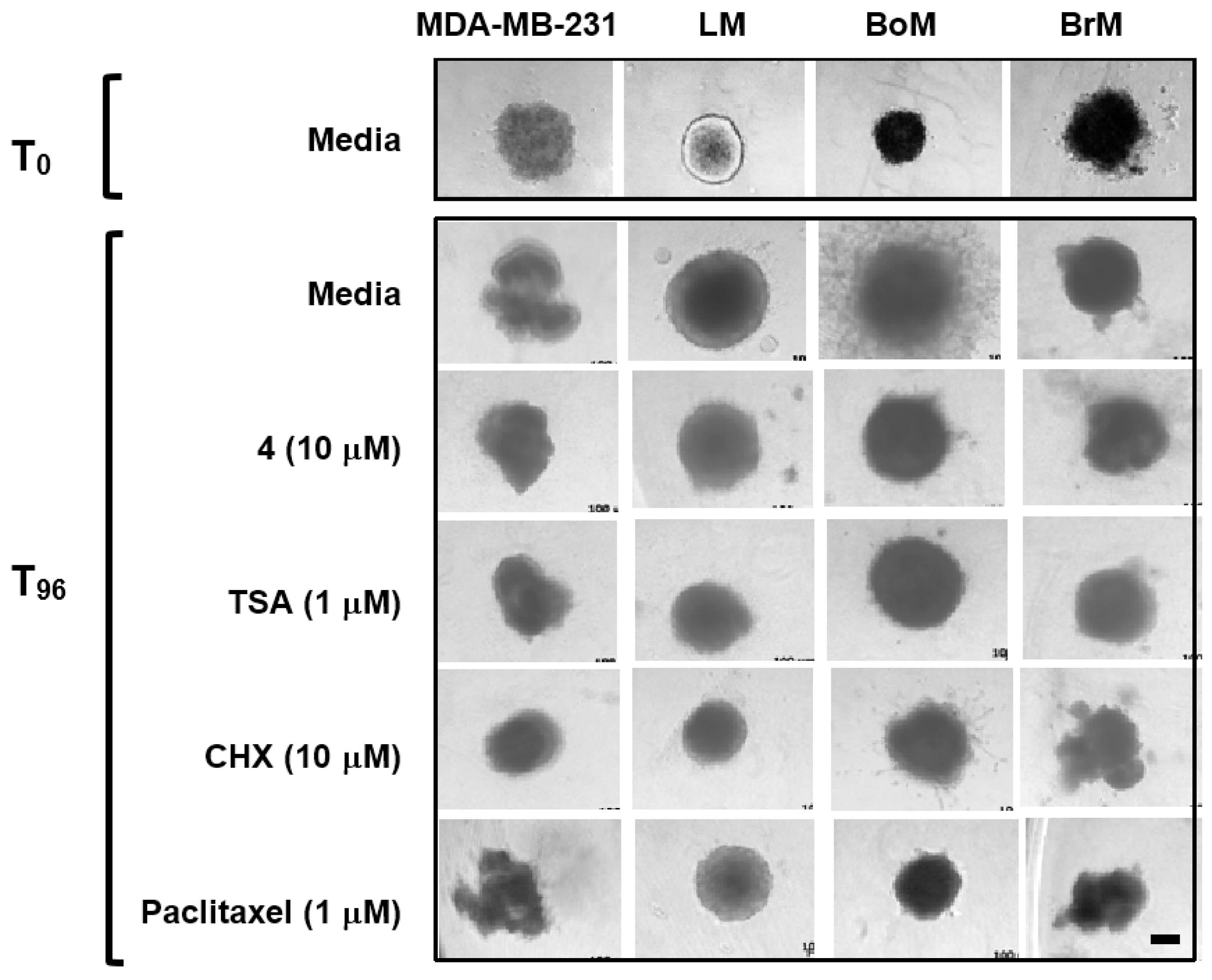
2.5. Psammaplin A and TSA Inhibit Tumor Cell Invasion
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Sponge Material, Extract Preparation, and Bioassay-Guided Isolation
4.3. Structural Data
4.4. T47D Cell-Based Reporter Assay
4.5. MDA-MB-435 Cell-Based HDAC Assay
4.6. Quantitative Real-Time RT-PCR and ELISA Assay
4.7. Cell Proliferation/Viability and Clonogenic Survival Assays
4.8. 3D Cell Invasion Assay
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Breast Cancer, Facts & Figures 2018. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2018/cancer-facts-and-figures-2018.pdf (accessed on 10 January 2018).
- Kang, Y.; Pantel, K. Tumor cell dissemination: Emerging biological insights from animal models and cancer patients. Cancer Cell 2013, 23, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Pantel, K.; Kang, Y. Tumor metastasis: moving new biological insights into the clinic. Nat. Med. 2013, 19, 1450–1464. [Google Scholar] [CrossRef] [PubMed]
- Vanharanta, S.; Massagué, J. Origins of metastatic traits. Cancer Cell 2013, 24, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Sethi, N.; Kang, Y. Unravelling the complexity of metastasis—molecular understanding and targeted therapies. Nat. Rev. Cancer 2011, 11, 735–748. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute at the National Institutes of Health. Clinical Trials Information for Patients and Caregivers; 2018. Available online: http://www.cancer.gov/clinicaltrials (accessed on 10 January 2018).
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Minn, A.J.; Kang, Y.; Serganova, I.; Gupta, G.P.; Giri, D.D.; Doubrovin, M.; Ponomarev, V.; Gerald, W.L.; Blasberg, R.; Massagué, J. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J. Clin. Investig. 2005, 115, 44–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minn, A.J.; Gupta, G.P.; Siegel, P.M.; Bos, P.D.; Shu, W.; Giri, D.D.; Viale, A.; Olshen, A.B.; Gerald, W.L.; Massagué, J. Genes that mediate breast cancer metastasis to lung. Nature 2005, 436, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Minn, A.J.; Kang, Y.; Siegel, P.M.; Serganova, I.; Cordón-Cardo, C.; Olshen, A.B.; Gerald, W.L.; Massagué, J. Identifying site-specific metastasis genes and functions. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Massagué, J. Genetic determinants of cancer metastasis. Nat. Rev. Genet. 2007, 8, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.J.; Gupta, G.P.; Padua, D.; Bos, P.; Nguyen, D.X.; Nuyten, D.; Kreike, B.; Zhang, Y.; Wang, Y.; Ishwaran, H.; et al. Lung metastasis genes couple breast tumor size and metastatic spread. Proc. Natl. Acad. Sci. USA 2007, 104, 6740–6745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, G.P.; Perk, J.; Acharyya, S.; de Candia, P.; Mittal, V.; Todorova-Manova, K.; Gerald, W.L.; Brogi, E.; Benezra, R.; Massagué, J. ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc. Natl. Acad. Sci. USA 2007, 104, 19506–19511. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Bos, P.D.; Zhang, X.H.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Piña, I.C.; Gautschi, J.T.; Wang, G.Y.; Sanders, M.L.; Schmitz, F.J.; France, D.; Cornell-Kennon, S.; Sambucetti, L.C.; Remiszewski, S.W.; Perez, L.B.; et al. Psammaplins from the sponge Pseudoceratina purpurea: inhibition of both histone deacetylase and DNA methyltransferase. J. Org. Chem. 2003, 68, 3866–3873. [Google Scholar] [CrossRef] [PubMed]
- Baud, M.G.; Leiser, T.; Haus, P.; Samlal, S.; Wong, A.C.; Wood, RJ.; Petrucci, V.; Gunaratnam, M.; Hughes, S.M.; Buluwela, L.; et al. Defining the mechanism of action and enzymatic selectivity of psammaplin A against its epigenetic targets. J. Med. Chem. 2012, 55, 1731–1750. [Google Scholar] [CrossRef] [PubMed]
- García, J.; Franci, G.; Pereira, R.; Benedetti, R.; Nebbioso, A.; Rodríguez-Barrios, F.; Gronemeyer, H.; Altucci, L.; de Lera, A.R. Epigenetic profiling of the antitumor natural product psammaplin A and its analogues. Bioorg. Med. Chem. 2011, 19, 3637–3649. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Semenza, G.L. Maintenance of redox homeostasis by hypoxia-inducible factors. Redox Biol. 2017, 13, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Hodges, T.W.; Hossain, C.F.; Kim, Y.-P.; Zhou, Y.-D.; Nagle, D.G. Molecular-targeted antitumor agents: The Saururus cernuus dineolignans manassantin B and 4-O-demethylmanassantin B are potent inhibitors of hypoxia-activated HIF-1. J. Nat. Prod. 2004, 67, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Veena, C.K.; Morgan, J.B.; Mohammed, K.A.; Jekabsons, M.B.; Nagle, D.G.; Zhou, Y.-D. Methylalpinumisoflavone inhibits hypoxia-inducible factor-1 (HIF-1) activation by simultaneously targeting multiple mechanisms. J. Biol. Chem. 2009, 284, 5859–5868. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.B.; Mahdi, F.; Liu, Y.; Coothankandaswamy, V.; Jekabsons, M.B.; Johnson, T.A.; Sashidhara, K.V.; Crews, P.; Nagle, D.G.; Zhou, Y.-D. The marine sponge metabolite mycothiazole: A novel prototype mitochondrial complex I inhibitor. Bioorg. Med. Chem. 2010, 18, 5988–5994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Semenza, G.L. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: Implications for models of hypoxia signal transduction. Blood 1993, 82, 3610–3615. [Google Scholar] [PubMed]
- Xia, M.; Huang, R.; Sun, Y.; Semenza, G.L.; Aldred, S.F.; Witt, K.L.; Inglese, J.; Tice, R.R.; Austin, C.P. Identification of chemical compounds that induce HIF-1α activity. Toxicol. Sci. 2009, 112, 153–163. [Google Scholar] [CrossRef]
- Befani, C.; Mylonis, I.; Gkotinakou, I.M.; Georgoulias, P.; Hu, C.J.; Simos, G.; Liakos, P. Cobalt stimulates HIF-1-dependent but inhibits HIF-2-dependent gene expression in liver cancer cells. Int. J. Biochem. Cell Biol. 2013, 45, 2359–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia-inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 and human disease: One highly involved factor. Genes Dev. 2000, 14, 1983–1991. [Google Scholar] [PubMed]
- VEGF/VEGFR Inhibitors; Drugs.com. 2018. Available online: https://www.drugs.com/drug-class/vegf-vegfr-inhibitors.html (accessed on 16 June 2018).
- National Institutes of Health/National Cancer Institute online. Angiogenesis Inhibitors; 2018. Available online: https://www.cancer.gov/about-cancer/treatment/types/immunotherapy/angiogenesis-inhibitors-fact-sheet#q3 (accessed on 16 June 2018).
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robles, A.J.; Du, L.; Cichewicz, R.H.; Mooberry, S.L. Maximiscin induces DNA damage, activates DNA damage response pathways, and has selective cytotoxic activity against a subtype of triple-negative breast cancer. J. Nat. Prod. 2016, 79, 1822–1827. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health/National Cancer Institute online. Romidepsin—Cancer Treatment, A to Z List of Cancer Drugs, updated 9 March 2018. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/romidepsin (accessed on 14 June 2018).
- National Institutes of Health/National Cancer Institute online. Vorinostat—Cancer Treatment, A to Z List of Cancer Drugs, updated 9 March 2018. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/vorinostat (accessed on 14 June 2018).
- Kim, T.H.; Kim, H.S.; Kang, Y.J.; Yoon, S.; Lee, J.; Choi, W.S.; Jung, J.H.; Kim, H.S. Psammaplin A induces Sirtuin 1-dependent autophagic cell death in doxorubicin-resistant MCF-7/adr human breast cancer cells and xenografts. Biochim. Biophys. Acta 2015, 1850, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Ratovitski, E.A. Tumor protein (TP)-p53 members as regulators of autophagy in tumor cells upon marine drug exposure. Mar. Drugs 2016, 14, e154. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Shan, Y.; Tao, L.; Liu, Y.; Zhu, Z.; Liu, Z.; Wu, Y.; Chen, W.; Wang, A.; Lu, Y. Diallyl trisulfides, a natural histone deacetylase inhibitor, attenuate HIF-1α synthesis, and decreases breast cancer metastasis. Mol. Carcinog. 2017, 56, 2317–2331. [Google Scholar] [CrossRef] [PubMed]
- McCloud, T.G. High-throughput extraction of plant, marine and fungal specimens for preservation of biologically active molecules. Molecules 2010, 15, 4526–4563. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Liu, Y.; Hong, J.; Lee, C.O.; Cho, H.; Kim, D.K.; Im, K.S.; Jung, J.H. New bromotyrosine derivatives from an association of two sponges, Jaspis wondoensis and Poecillastra wondoensis. J. Nat. Prod. 2003, 66, 1495–1498. [Google Scholar] [CrossRef] [PubMed]
- Tabudravu, J.N.; Eijsink, V.G.H.; Gooday, G.W.; Jaspars, M.; Komander, D.; Legg, M.; Synstad, B.; van Aalten, D.M.F. Psammaplin A, a chitinase inhibitor isolated from the Fijian marine sponge Aplysinella rhax. Bioorg. Med. Chem. 2002, 10, 1123–1128. [Google Scholar] [CrossRef]
- Rodriguez, A.D.; Akee, R.K.; Scheuer, P.J. Two bromotyrosine-cysteine derived metabolites from a sponge. Tetrahedron Lett. 1987, 28, 4989–4992. [Google Scholar] [CrossRef]
- Hossain, C.F.; Kim, Y.-P.; Baerson, S.R.; Zhang, L.; Bruick, R.K.; Mohammed, K.A.; Agarwal, A.K.; Nagle, D.G.; Zhou, Y.-D. Saururus cernuus lignans--potent small molecule inhibitors of hypoxia-inducible factor-1. Biochem. Biophys. Res. Comm. 2005, 333, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-D.; Kim, Y.-P.; Li, X.-C.; Baerson, S.R.; Agarwal, A.K.; Hodges, T.W.; Ferreira, D.; Nagle, D.G. Hypoxia-inducible factor-1 activation by (-)-epicatechin gallate: potential adverse effects of cancer chemoprevention with high-dose green tea extracts. J. Nat. Prod. 2004, 67, 2063–2069. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) | 95% CI (µM) 1 |
|---|---|---|
| 1 | 0.257 | 0.157–0.420 |
| 2 | 0.019 | 0.012–0.028 |
| 3 | 0.038 | 0.024–0.061 |
| 4 | 0.037 | 0.025–0.055 |
| 5 | 0.948 | 0.586–1.532 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.-D.; Li, J.; Du, L.; Mahdi, F.; Le, T.P.; Chen, W.-L.; Swanson, S.M.; Watabe, K.; Nagle, D.G. Biochemical and Anti-Triple Negative Metastatic Breast Tumor Cell Properties of Psammaplins. Mar. Drugs 2018, 16, 442. https://doi.org/10.3390/md16110442
Zhou Y-D, Li J, Du L, Mahdi F, Le TP, Chen W-L, Swanson SM, Watabe K, Nagle DG. Biochemical and Anti-Triple Negative Metastatic Breast Tumor Cell Properties of Psammaplins. Marine Drugs. 2018; 16(11):442. https://doi.org/10.3390/md16110442
Chicago/Turabian StyleZhou, Yu-Dong, Jun Li, Lin Du, Fakhri Mahdi, Thuy P. Le, Wei-Lun Chen, Steven M. Swanson, Kounosuke Watabe, and Dale G. Nagle. 2018. "Biochemical and Anti-Triple Negative Metastatic Breast Tumor Cell Properties of Psammaplins" Marine Drugs 16, no. 11: 442. https://doi.org/10.3390/md16110442