
Study on the Antifibrotic Effects of Recombinant Shark Hepatical Stimulator Analogue (r-sHSA) in Vitro and in Vivo

Abstract
:1. Introduction
2. Results
2.1. r-sHSA Inhibits Fibroblasts in Vitro

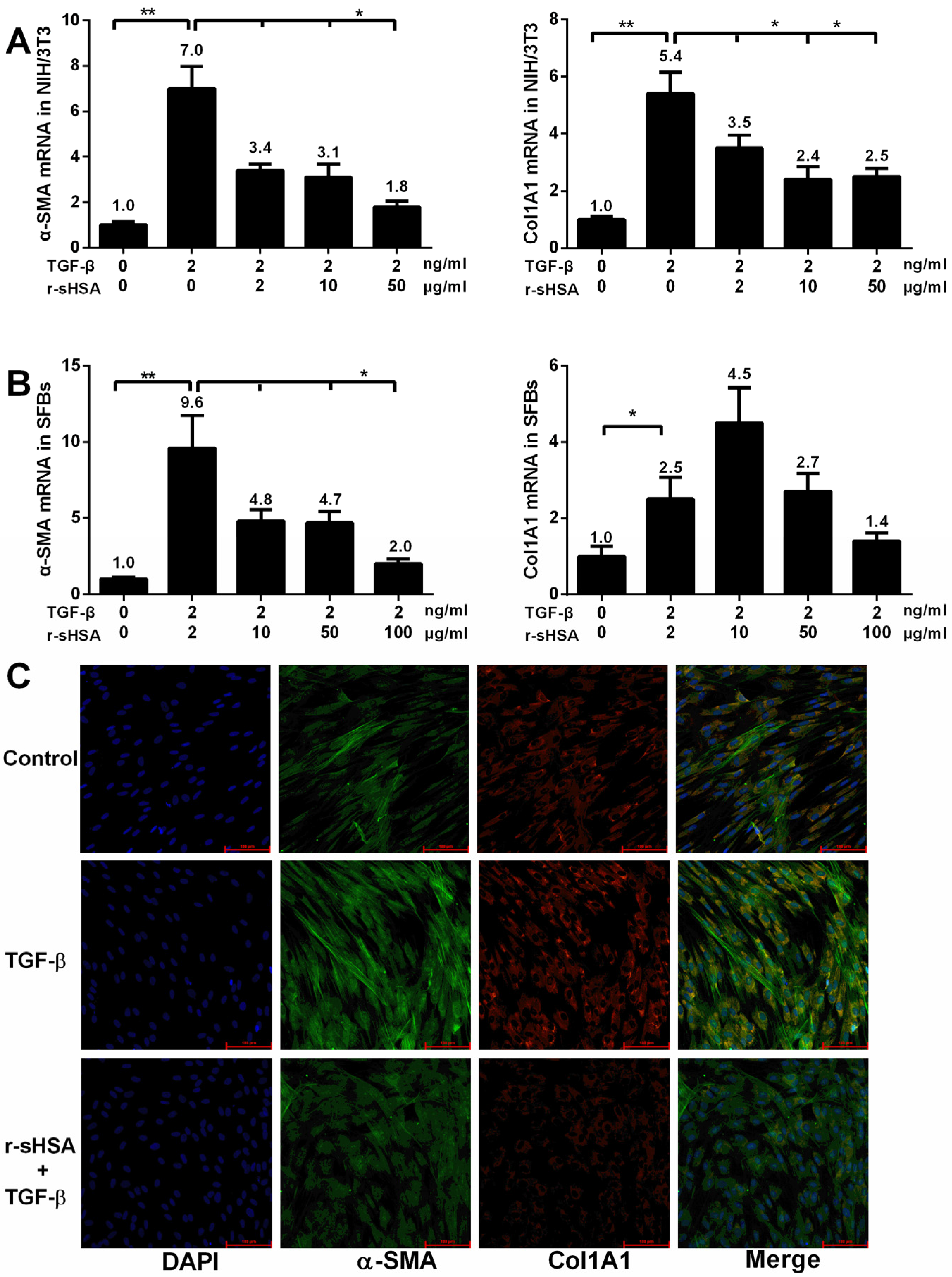{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward (5′-3′) | Reverse (5′-3′) | Accession Number |
|---|---|---|---|
| Mouse 18S rRNA | CCCCATGAACGAGGGAATT | GGGACTTAATCAACGCAAGCTT | NM_08324.3 |
| Mouse COL1A1 | TGGTCCCAAAGGTTCTCCTGGT | TTAGGTCCAGGGAATCCCATCACA | NM_053304.1 |
| Mouse α-SMA | CTATAACCGGAACTTCTGCCAG | CTGCTCTGTGTCAGGTGTG | NM_031004.2 |
| Human COL1A1 | GCTGGTGTGATGGGATTC | GGGAACACCTCGCTCT | NM_000088 |
| Human α-SMA | CAGGGCTGTTTTCCCATCCAT | GCCATGTTCTATCGGGTACTTC | NM_001613 |
| Human 18S rRNA | CCCCATGAACGAGGGAATT | GGGACTTAATCAACGCAAGCTT | NR_003286 |
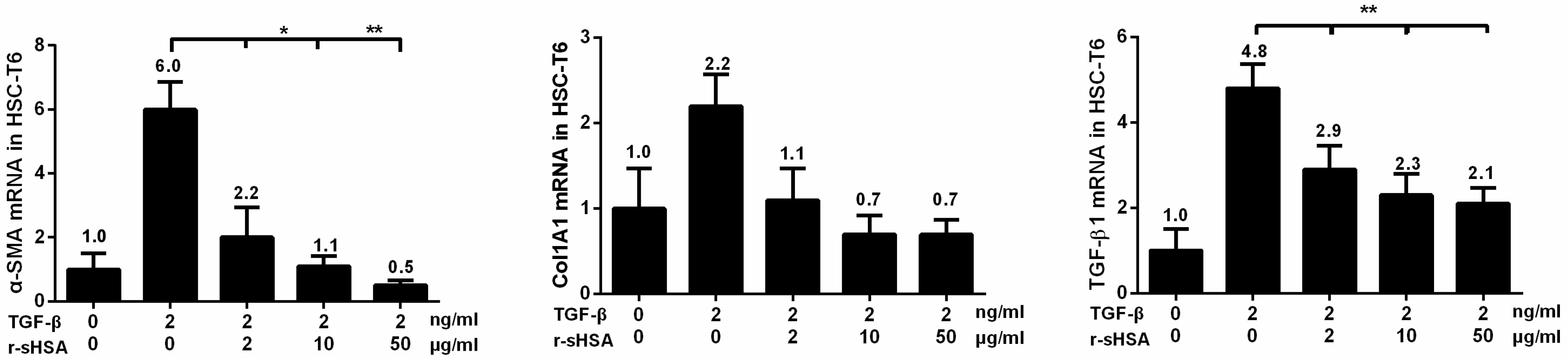
2.2. r-sHSA Attenuates TGF-β-Induced HSCs Activation
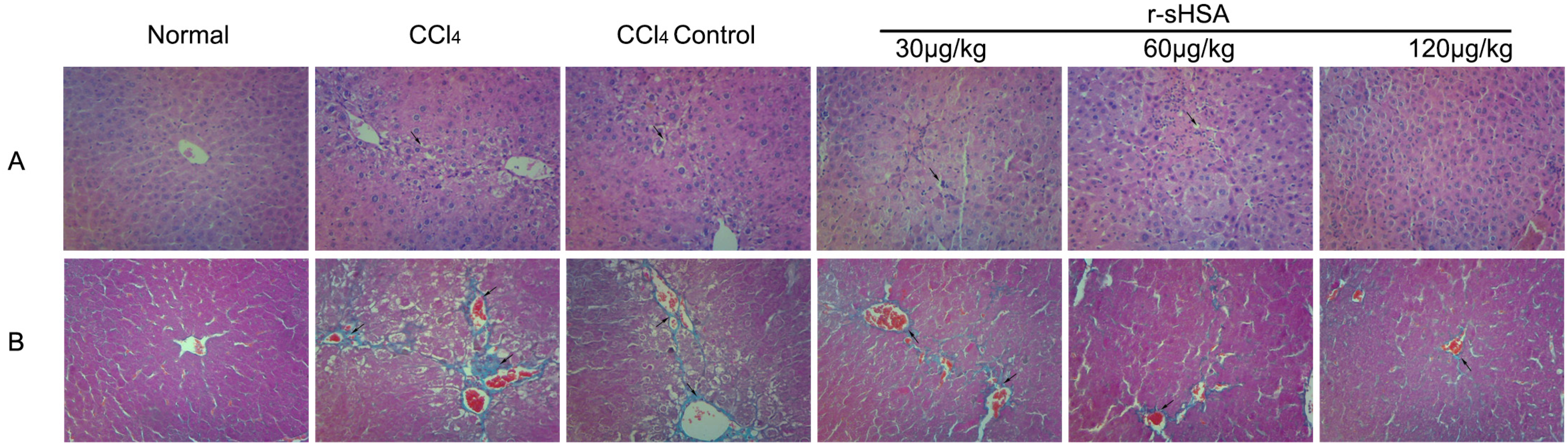
2.3. r-sHSA Ameliorates Liver Injury Induced by CCl4


2.4. r-sHSA Alleviates CCl4-Induced Hepatic Fibrosis
| Normal | CCl4 | CCl4 control | r-sHSA | |||
|---|---|---|---|---|---|---|
| 30 μg/kg | 60 μg/kg | 120 μg/kg | ||||
| ALT (IU/L) | 40.19 ± 14.47 **++ | 166.80 ± 37.12 ++ | 97.47 ± 32.61 ** | 89.57 ± 16.72 ** | 74.37 ± 32.39 ** | 72.53 ± 27.77 ** |
| AST (IU/L) | 65.75 ± 9.86 **++ | 244.80 ± 61.55 ++ | 107.46 ± 32.37 ** | 89.24 ± 11.64 ** | 78.85 ± 29.03 **+ | 75.35 ± 23.07 **+ |
| ALP(IU/L) | 38.33 ± 3.20 ** | 57.71 ± 6.97 + | 41.06 ± 10.32 * | 40.81 ± 6.83 ** | 43.54 ± 4.85 ** | 40.27 ± 2.41 ** |
| HA(ng/mL) | 74.17 ± 10.70 **++ | 133.10 ± 15.31 ++ | 101.10 ± 9.61 ** | 95.30 ± 9.73 ** | 83.60 ± 4.57 **++ | 85.66 ± 7.51 **+ |
| TIMP-1 (ng/mL) | 4.68 ± 0.44 **+ | 8.53 ± 0.71 ++ | 5.63 ± 0.67 ** | 6.01 ± 0.84 ** | 5.49 ± 0.52 ** | 4.59 ± 0.41 **+ |
| Hyp (μg/g liver) | 136.92 ± 14.43 **++ | 487.39 ± 31.83 + | 402.99 ± 70.56 * | 387.07 ± 37.46 ** | 312.98 ± 47.06 **+ | 199.02 ± 18.41 **++ |
| Collagen III (ug/g protein) | 0.35 ± 0.06 **++ | 0.94 ± 0.18 + | 0.71 ± 0.11 * | 0.55 ± 0.14 ** | 0.49 ± 0.05 **+ | 0.47 ± 0.11 **+ |
| Normal | CCl4 | CCl4 control | r-sHSA | |||
|---|---|---|---|---|---|---|
| 30 μg/kg | 60 μg/kg | 120 μg/kg | ||||
| MDA (nM/mg protein) | 5.03 ± 1.59 * | 7.88 ± 1.34 + | 5.94 ± 1.20 * | 5.73 ± 0.82 * | 4.74 ± 0.86 ** | 5.13 ± 0.83 ** |
| GSH (mg/g protein) | 3.80 ± 0.56 ** | 2.66 ± 0.30 + | 3.30 ± 0.53 * | 4.49 ± 1.20 ** | 6.11 ± 1.21 **++ | 6.89 ± 0.87 **++ |
| TAOC (U/mg protein) | 1.57 ± 0.26 ** | 1.02 ± 0.22 + | 1.37 ± 0.13 * | 1.49 ± 0.25 ** | 1.67 ± 0.41 * | 1.67 ± 0.22 *+ |
| Normal | CCl4 | CCl4 control | r-sHSA | |||
|---|---|---|---|---|---|---|
| 30 μg/kg | 60 μg/kg | 120 μg/kg | ||||
| Fibrosis score | 0 ± 0 | 3.42 ± 0.76 *++ | 2.33 ± 0.94 ** | 2.00 ± 0.82 ** | 1.83 ± 0.80 ** | 0.83 ± 0.69 **++ |
| Hepatocyte necrosis | 0 ± 0 | 1.67 ± 0.62 *+ | 1.08 ± 0.64 * | 1.00 ± 0.71 * | 0.58 ± 0.49 ** | 0.17 ± 0.37 **++ |

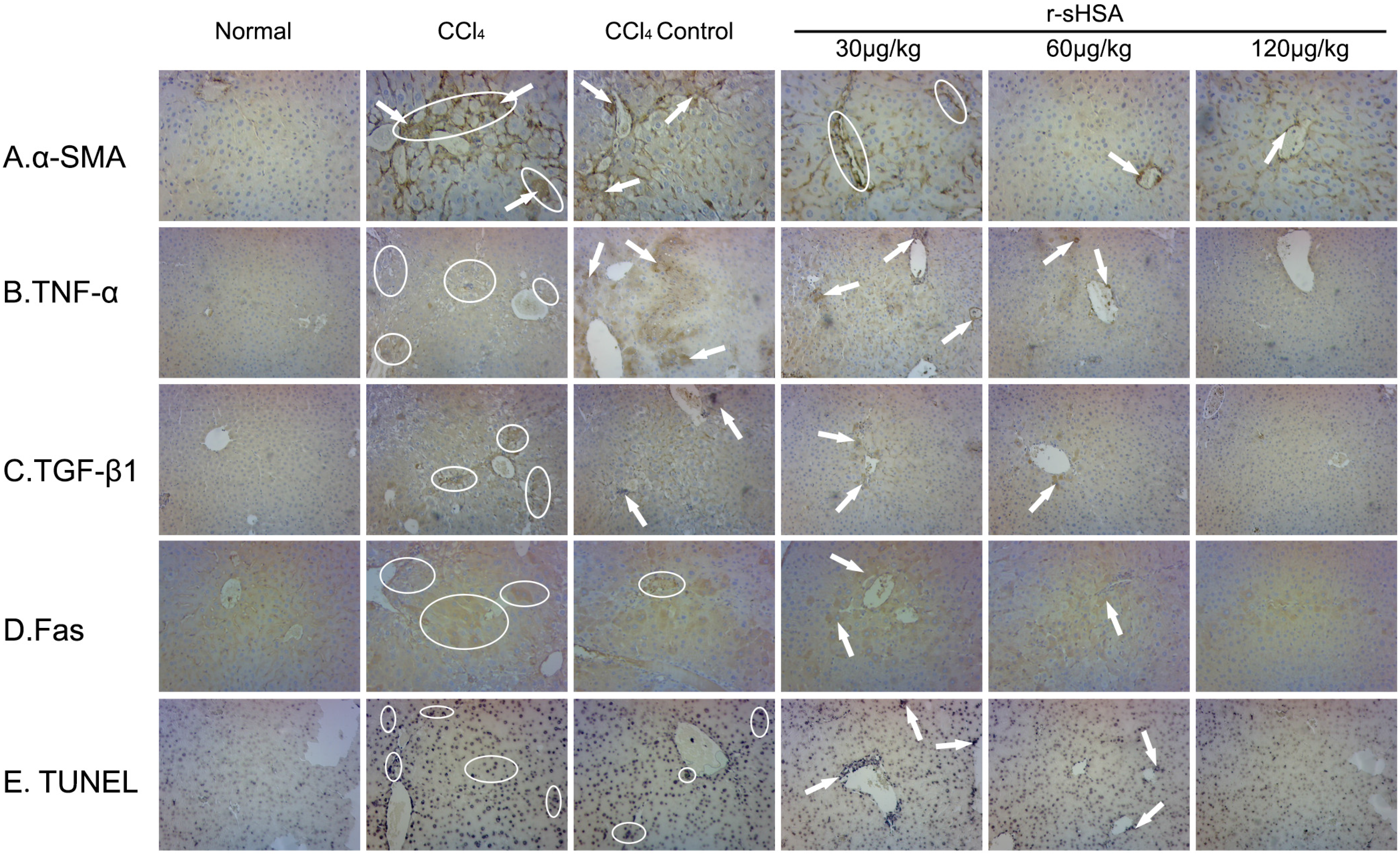
2.5. r-sHSA Inhibits HSCs Activation and Inflammation
2.6. r-sHSA Moderates Hepatocyte Apoptosis

3. Discussion
4. Experimental Section
4.1. Preparation of r-sHSA
4.2. Cells and Cell Culture
4.3. Antifibrotic Assay in Vitro
4.4. Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT-PCR)
4.5. Confocal Immunofluorescence Microscopy
4.6. Mice and Hepatic Fibrosis Model
4.7. Biochemical Analysis
4.8. Histopathologic Evaluation
4.9. TUNEL and Immunohistochemical Assay
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| r-sHSA | recombinant shark Hepatical Stimulator Analogue |
| ECM | extracellular matrix |
| HSCs | hepatic stellate cells |
| ROS | reactive oxygen species |
| α-SMA | α-smooth muscle actin |
| TNF-α | tumor necrosis factor alpha |
| TGF-β | transforming growth factor beta |
| CCl4 | carbon tetrachloride |
| SFBs | human skin fibroblasts |
| FBS | fetal bovine serum |
| ALT | alanine aminotransferase |
| AST | aspartate aminotransferase |
| ALP | alkaline phosphatase |
| HA | hyaluronic acid |
| TIMP-1 | tissue inhibitor of metalloproteinases-1 |
| Hyp | hydroxyproline |
| MDA | malonaldehyde |
| TAOC | total antioxidant capacity |
| GSH | glutathione |
| i-NOS | inducible nitric oxide synthase |
| Col1A1 | alpha-1 type I collagen |
| TUNEL | terminal dexynucleotidyl transferase(TdT)-mediated dUTP nick end labeling |
References
- Zakim, D.; Boyer, T.D.; Manns, M.P.; Sanyal, A.J. Zakim and Boyer’s Hepatology: A Textbook of Liver Disease, 6th ed.; Elsevier Health Sciences: Kidlington, Oxford, UK, 2011. [Google Scholar]
- Bataller, R.; Brenner, D.A. Hepatic Stellate Cells as a Target for the Treatment of Liver Fibrosis. Semin. Liver Dis. 2001, 21, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Invest. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Reeves, H.L.; Friedman, S.L. Activation of hepatic stellate cells—A key issue in liver fibrosis. Front. Biosci. 2002, 7, 808–826. [Google Scholar] [CrossRef]
- Palmes, D.; Spiegel, H.-U. Animal models of liver regeneration. Biomaterials 2004, 25, 1601–1611. [Google Scholar] [CrossRef]
- Heron, M. Deaths: Leading causes for 2004. Natl. Vital Stat. Rep. 2007, 56, 1–96. [Google Scholar] [PubMed]
- Benyon, R.; Iredale, J. Is liver fibrosis reversible? Gut 2000, 46, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P. Models of liver fibrosis: Exploring the dynamic nature of inflammation and repair in a solid organ. J. Clin. Invest. 2007, 117, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Atzori, L.; Poli, G.; Perra, A. Hepatic stellate cell: A star cell in the liver. Int. J. Biochem. Cell Biol. 2009, 41, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Svegliati-Baroni, G.; Saccomanno, S.; Goor, H.; Jansen, P.; Benedetti, A.; Moshage, H. Involvement of reactive oxygen species and nitric oxide radicals in activation and proliferation of rat hepatic stellate cells. Liver 2001, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Gressner, O.A.; Weiskirchen, R.; Gressner, A.M. Evolving concepts of liver fibrogenesis provide new diagnostic and therapeutic options. Comp. Hepatol. 2007, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, B.; Xi, T.; Lv, Z.; Li, Q.; Wang, Y.; Wang, H.; Wu, W. Cloning and sequence analysis of cDNA fragment of shark hepatic stimulator substance’s similarity. Chin. J. Nat. Med. 2003, 1, 111–115. [Google Scholar]
- Wang, Y.; Bian, S.; Zhang, T.; Zhao, Y.; Wu, W.; Ye, B. Expressing and bioactivity analysis of shark hepatic stimulate substance analogue in Escherichia coli. Mar. Sci. 2004, 28, 37–41. [Google Scholar]
- Wang, Y.; Zhao, Y.; Ye, B.; Eng, Y.; Wu, W.; Wang, M. Effect of recombinant hepatical stimulator substance analogue on CCl4-induced acute liver injury in mice. J. Chin. Pharm. Univ. 2005, 36, 368–372. [Google Scholar]
- Zhang, X.; Hang, Y.; Wang, J.; Ding, X.; Wang, Y.; Ye, B. Protective effect of recombinant hepatical stimulator analogue on Concanavalin A-induced acute liver injury in mice. J. Chin. Pharm. Univ. 2011, 42, 375–379. [Google Scholar]
- Drotman, R.; Lawhorn, G. Serum enzymes as indicators of chemically induced liver damage. Drug Chem. Toxicol. 1978, 1, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Ozer, J.; Ratner, M.; Shaw, M.; Bailey, W.; Schomaker, S. The current state of serum biomarkers of hepatotoxicity. Toxicology 2008, 245, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Robino, G. Oxidative stress-related molecules and liver fibrosis. J. Hepatol. 2001, 35, 297–306. [Google Scholar] [CrossRef]
- Gillette, J.R.; Mitchell, J.R.; Brodie, B.B. Biochemical mechanisms of drug toxicity. Annu. Rev. Pharmacol. 1974, 14, 271–288. [Google Scholar] [CrossRef]
- Berger, M.L.; Bhatt, H.; Combes, B.; Estabrook, R.W. CCl4-induced toxicity in isolated hepatocytes: The importance of direct solvent injury. Hepatology 1986, 6, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Recknagel, R.O.; Glende, E.A.; Dolak, J.A.; Waller, R.L. Mechanisms of carbon tetrachloride toxicity. Pharmacol. Ther. 1989, 43, 139–154. [Google Scholar] [CrossRef]
- Brattin, W.J.; Glende, E.A.; Recknagel, R.O. Pathological mechanisms in carbon tetrachloride hepatotoxicity. J. Free Radic. Biol. Med. 1985, 1, 27–38. [Google Scholar] [CrossRef]
- Basu, S. Carbon tetrachloride-induced lipid peroxidation: Eicosanoid formation and their regulation by antioxidant nutrients. Toxicology 2003, 189, 113–127. [Google Scholar] [CrossRef]
- Ha, H.; Shin, H.; Feitelson, M.; Yu, D. Oxidative stress and antioxidants in hepatic pathogenesis. World J. Gastroenterol. 2010, 16, 6035–6043. [Google Scholar] [CrossRef] [PubMed]
- Apostolova, N.; Victor, V.M. Molecular strategies for targeting antioxidants to mitochondria: Therapeutic implications. Antioxid. Redox Signaling 2015, 22, 686–729. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.-F.; DeLellis, R. Reversibility of hepatic fibrosis in autoimmune hepatitsis. Ann. Intern. Med. 1997, 127, 981. [Google Scholar] [CrossRef] [PubMed]
- Wanless, I.R.; Nakashima, E.; Sherman, M. Regression of human cirrhosis. Morphologic features and the genesis of incomplete septal cirrhosis. Arch. Pathol. Lab. Med. 2000, 124, 1599–1607. [Google Scholar] [PubMed]
- Abdel-Aziz, G.; Lebeau, G.; Rescan, P.; Clement, B.; Rissel, M.; Deugnier, Y.; Campion, J.; Guillouzo, A. Reversibility of hepatic fibrosis in experimentally induced cholestasis in rat. Am. J. Pathol. 1990, 137, 1333–1342. [Google Scholar] [PubMed]
- Iredale, J.; Benyon, R.; Pickering, J.; McCullen, M.; Northrop, M.; Pawley, S.; Hovell, C.; Arthur, M. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J. Clin. Invest. 1998, 102, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Fracanzani, A.L.; Valenti, L.; Bugianesi, E.; Andreoletti, M.; Colli, A.; Vanni, E.; Bertelli, C.; Fatta, E.; Bignamini, D.; Marchesini, G. Risk of severe liver disease in nonalcoholic fatty liver disease with normal aminotransferase levels: A role for insulin resistance and diabetes. Hepatology 2008, 48, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Domitrović, R.; Jakovac, H.; Tomac, J.; Šain, I. Liver fibrosis in mice induced by carbon tetrachloride and its reversion by luteolin. Toxicol. Appl. Pharmacol. 2009, 241, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 2000, 50, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Landriscina, M.; Maddalena, F.; Laudiero, G.; Esposito, F. Adaptation to oxidative stress, chemoresistance, and cell survival. Antioxid. Redox Signaling 2009, 11, 2701–2716. [Google Scholar] [CrossRef] [PubMed]
- Afdhal, N.H.; Nunes, D. Evaluation of liver fibrosis: A concise review. Am. J. Gastroenterol. 2004, 99, 1160–1174. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, S.; Parsons, C.J.; Rippe, R.A. Mechanisms of liver fibrosis. Clin. Chim. Acta 2006, 364, 33–60. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Flier, J.S.; Underhill, L.H.; Friedman, S.L. The cellular basis of hepatic fibrosis—Mechanisms and treatment strategies. N. Engl. J. Med. 1993, 328, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Lotersztajn, S.; Julien, B.; Teixeira-Clerc, F.; Grenard, P.; Mallat, A. Hepatic fibrosis: Molecular mechanisms and drug targets. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 605–628. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Roll, F.J.; Boyles, J.; Bissell, D.M. Hepatic lipocytes: The principal collagen-producing cells of normal rat liver. Proc. Natl. Acad. Sci. USA 1985, 82, 8681–8685. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Forbes, S.J. Hepatic fibrogenesis: From within and outwith. Toxicology 2008, 254, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Cytokines and Fibrogenesis. Semin. Liver Dis. 1999, 19, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H. Cytokine regulation of hepatic stellate cells in liver fibrosis. Alcohol. Clin. Exp. Res. 1999, 23, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 2000, 275, 2247–2250. [Google Scholar] [CrossRef] [PubMed]
- Wasmuth, H.E.; Tacke, F.; Trautwein, C. Chemokines in liver inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Knittel, T.; Mehde, M.; Kobold, D.; Saile, B.; Dinter, C.; Ramadori, G. Expression patterns of matrix metalloproteinases and their inhibitors in parenchymal and non-parenchymal cells of rat liver: Regulation by TNF-α and TGF-β1. J. Hepatol. 1999, 30, 48–60. [Google Scholar] [CrossRef]
- Gressner, A.M.; Weiskirchen, R.; Breitkopf, K.; Dooley, S. Roles of TGF-beta in hepatic fibrosis. Front. Biosci. 2002, 7, d793–d807. [Google Scholar] [CrossRef] [PubMed]
- Gressner, A.; Weiskirchen, R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-β as major players and therapeutic targets. J. Cell. Mol. Med. 2006, 10, 76–99. [Google Scholar] [CrossRef] [PubMed]
- Breitkopf, K.; Godoy, P.; Ciuclan, L.; Singer, M.; Dooley, S. TGF-beta/smad signaling in the injured liver. Z. Gastroenterol. 2006, 44, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Barnard, J.A.; Lyons, R.M.; Moses, H.L. The cell biology of transforming growth factor β. Biochim. Biophy. Acta Rev. Cancer 1990, 1032, 79–87. [Google Scholar] [CrossRef]
- Overall, C.M.; Wrana, J.; Sodek, J. Independent regulation of collagenase, 72-kDa progelatinase, and metalloendoproteinase inhibitor expression in human fibroblasts by transforming growth factor-beta. J. Biol. Chem. 1989, 264, 1860–1869. [Google Scholar] [PubMed]
- Domitrović, R.; Jakovac, H. Antifibrotic activity of anthocyanidin delphinidin in carbon tetrachloride-induced hepatotoxicity in mice. Toxicology 2010, 272, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pinzani, M.; Rombouts, K. Liver fibrosis: From the bench to clinical targets. Dig. Liver Dis. 2004, 36, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Kovalovich, K.; DeAngelis, R.A.; Li, W.; Furth, E.E.; Ciliberto, G.; Taub, R. Increased toxin-induced liver injury and fibrosis in Interleukin-6–Deficient mice. Hepatology 2000, 31, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Canbay, A.; Friedman, S.; Gores, G.J. Apoptosis: The nexus of liver injury and fibrosis. Hepatology 2004, 39, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Canbay, A.; Higuchi, H.; Bronk, S.F.; Taniai, M.; Sebo, T.J.; Gores, G.J. Fas enhances fibrogenesis in the bile duct ligated mouse: A link between apoptosis and fibrosis. Gastroenterology 2002, 123, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.; Gores, G. Apoptosis: A mechanism of acute and chronic liver injury. Gut 2005, 54, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Pan, Z.; Li, H.; Wang, Y.; Zheng, H.; Wu, W. High cell-density fermentation of shark hepatical stimulator analogue in Escherichia coli. Chin. J. Biotechnol. 2009, 25, 1371. [Google Scholar]
- Varga, J.; Rosenbloom, J.; Jimenez, S. Transforming growth factor beta (TGF beta) causes a persistent increase in steady-state amounts of type I and type III collagen and fibronectin mRNAs in normal human dermal fibroblasts. Biochem. J. 1987, 247, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Chen, S.; Varga, J. Modulation of endogenous smad expression in normal skin fibroblasts by transforming growth factor-β. Exp. Cell Res. 2000, 258, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Kim, J.; Kim, H.; Park, I.-S.; Kim, I.Y.; Yang, B. Tyrosine 740 phosphorylation of discoidin domain receptor 2 by Src stimulates intramolecular autophosphorylation and Shc signaling complex formation. J. Biol. Chem. 2005, 280, 39058–39066. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Liu, L.; Yang, Y.; Wei, J.; Bhattacharyya, S.; Summer, R.; Ye, B.; Varga, J. Adiponectin has potent anti-fibrotic effects mediated via amp kinase: Novel target for fibrosis therap. Arthritis Rheum. 2012, 64, S967–S968. [Google Scholar]
- Wang, Y.; Zhang, M.; Wang, C.; Ye, B.; Hua, Z. Molecular cloning of the alpha subunit of complement component C8 (CpC8α) of whitespotted bamboo shark (Chiloscyllium plagiosum). Fish Shellfish Immunol. 2013, 35, 1993–2000. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, I.A.; Farrell, G.C.; Schriemer, R.; Robertson, G.R. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J. Hepatol. 2002, 37, 206–213. [Google Scholar] [CrossRef]
- Scheuer, P.J. Classification of chronic viral hepatitis: A need for reassessment. J. Hepatol. 1991, 13, 372–374. [Google Scholar] [CrossRef]
- Wang, L.; Cheng, D.; Wang, H.; Di, L.; Zhou, X.; Xu, T.; Yang, X.; Liu, Y. The hepatoprotective and antifibrotic effects of saururus chinensis against carbon tetrachloride induced hepatic fibrosis in rats. J. Ethnopharmacol. 2009, 126, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Chong, L.; Hsu, Y.; Chiu, Y.; Yang, K.; Huang, Y. Anti-fibrotic effects of thalidomide on hepatic stellate cells and dimethylnitrosamine-intoxicated rats. J. Biomed. Sci. 2006, 13, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Takiya, S.; Tagaya, T.; Takahashi, K.; Kawashima, H.; Kamiya, M.; Fukuzawa, Y.; Kobayashi, S.; Fukatsu, A.; Katoh, K.; Kakumu, S. Role of transforming growth factor beta 1 on hepatic regeneration and apoptosis in liver diseases. J. Clin. Pathol. 1995, 48, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhang, X.; Yang, Y.; Yang, X.; Ye, B. Study on the Antifibrotic Effects of Recombinant Shark Hepatical Stimulator Analogue (r-sHSA) in Vitro and in Vivo. Mar. Drugs 2015, 13, 5201-5218. https://doi.org/10.3390/md13085201
Wang Y, Zhang X, Yang Y, Yang X, Ye B. Study on the Antifibrotic Effects of Recombinant Shark Hepatical Stimulator Analogue (r-sHSA) in Vitro and in Vivo. Marine Drugs. 2015; 13(8):5201-5218. https://doi.org/10.3390/md13085201
Chicago/Turabian StyleWang, Ying, Xiaoyuan Zhang, Yang Yang, Xiaohong Yang, and Boping Ye. 2015. "Study on the Antifibrotic Effects of Recombinant Shark Hepatical Stimulator Analogue (r-sHSA) in Vitro and in Vivo" Marine Drugs 13, no. 8: 5201-5218. https://doi.org/10.3390/md13085201