Bioproduction of Chitooligosaccharides: Present and Perspectives

Abstract
:1. Introduction

2. Bioproduction of Chitin Oligosaccharides and Its Monomer
2.1. N-Acetyl-β-d-glucosamine

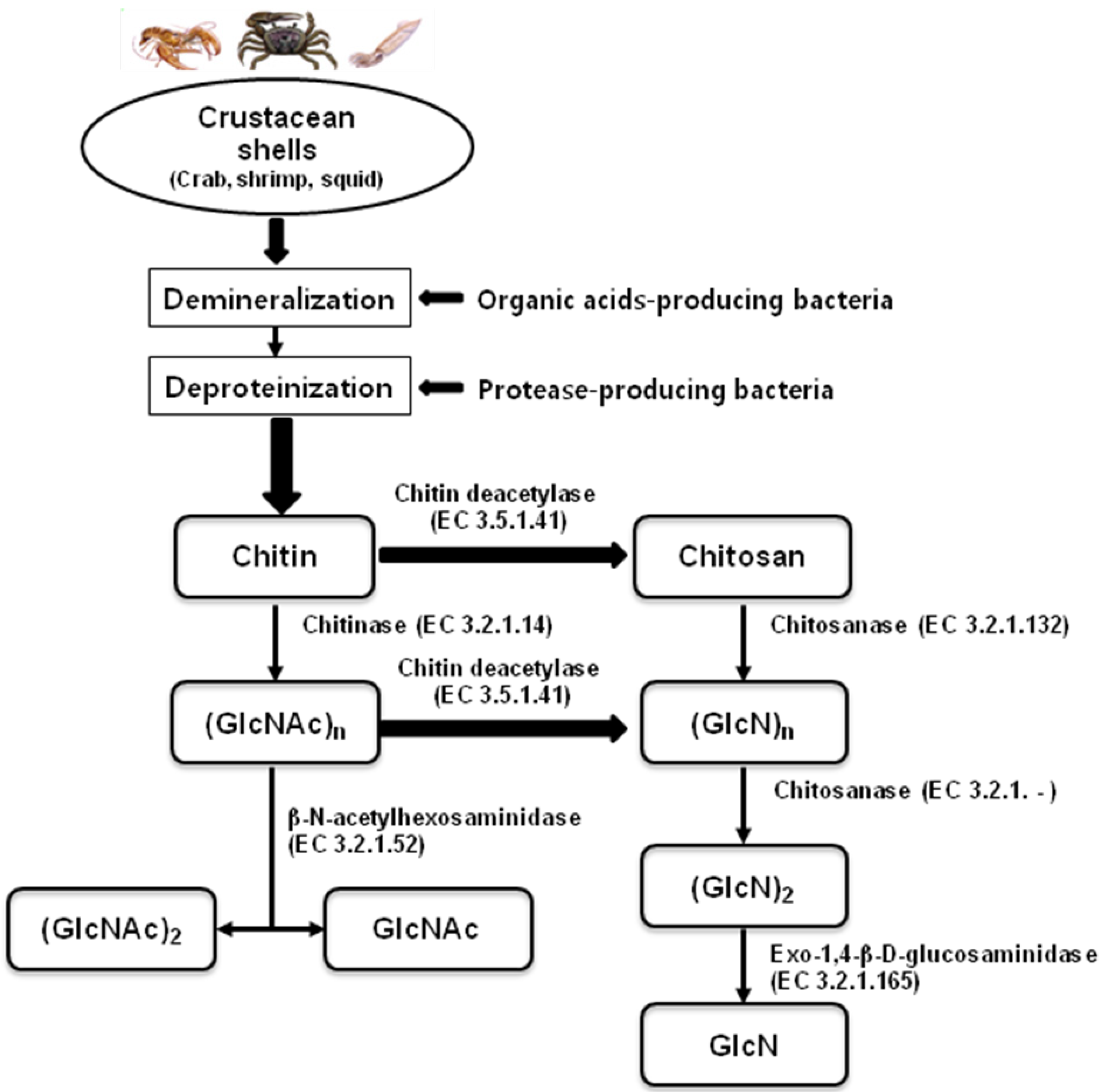{kind=link}
| Chitinsource | Enzyme Source | Enzyme | Mol. Wt. | Condition | Product & Yield | Analysis | Reference |
|---|---|---|---|---|---|---|---|
| Swollen chitin | Aeromonas sp. GJ-18 | Crude enzyme | - | 40 °C, 9 days | GlcNAc 94.9% | HPLC | [48] |
| NH2P-50 4E | |||||||
| Swollen chitin | Aeromonas sp. GJ-18 | Crude enzyme | - | 45 °C, 5 days | GlcNAc 74% | HPLC | [49] |
| (GlcNAc)2 4.8% | |||||||
| 55 °C, 5 days | GlcNAc 3.9% | NH2P-50 4E | |||||
| (GlcNAc)2 34.7% | |||||||
| α-Chitin | Aeromonas sp. GJ-18 | Crude enzyme | - | Preincubation (50 °C, 60 min) 45 °C, 7 days | (GlcNAc)2 78.9% | HPLC | [50] |
| β-Chitin | (GlcNAc)2 56.6% | NH2P-50 4E | |||||
| α-Chitin | A. hydrophila H-2330 | Crude enzyme | - | 17 °C, 10 days | GlcNAc 64~77% | HPLC | [44] |
| (Flake & powder) | NH2P-50 | ||||||
| Chitin | A. hydrophila SUWA-9 | Chitinase | - | 37 °C, overnight | (GlcNAc)2~(GlcNAc)5 | TLC | [47] |
| GlcNAc | |||||||
| Chitosan (60% DD) | Bacillus cereus TKU027 | Chitinase | 65/63 kDa | 37 °C, 2 h 30 °C, 2 days | (GlcNAc/GlcN) DP 4~9 | MALDI-TOF MS | [44] |
| Culture supernatant | (GlcNAc/GlcN) DP 2~5 | ||||||
| β-Chitin | Bacillus cereus TKU022 | Chitosanase | 44 kDa | 37 °C, 2 days | (GlcNAc)2, (GlcNAc)4 | HPLC | [51] |
| (GlcNAc)5, (GlcNAc)6 | |||||||
| Colloidal chitin | Bacillus thuringiensis subsp. pakistani | Exochitinase | 66/60/47/32 kDa | 37 °C, 24 h | GlcNAc | TLC | [45] |
| α-Chitin | Burkholderia cepacia TU09 | Chitinase | 37 °C, 7 days 37 °C, 1 days | GlcNAc 85% | HPLCNH2P-50 | [40] | |
| β-Chitin | GlcNAc 85% | ||||||
| α-Chitin | Bacillus licheniformis SK-1 | Chitinase | 37 °C, 6 days | GlcNAc41% | HPLCNH2P-50 | ||
| β-Chitin | GlcNAc75% | ||||||
| Swollen chitin | Paenibacillus illinoisensis KJA-424 | Chitinase | 38/54/63 kDa | 37 °C, 24 h | GlcNAc 62.2% | HPLC | [7] |
| Chitin | Trichoderma reesei | Cellulases &β-glucanases | - | 37 °C, 10 days | GlcNAc 86% | TLC/HPLC Separon SGX NH2 | [42] |
| α-Chitin | Trichoderma viride | Cellulase | - | 37 °C, 3 days | GlcNAc 16% | HPLC/NMR | [41] |
| Acremonium cellulolyticus | GlcNAc 22% | ||||||
| β-Chitin | Aspergillus niger | Cellulose & lipase | - | 37 °C, 4 days | GlcNAc 61% | HPLC | [43] |
| NH2P-50 | |||||||
| Chitin | Thermococcus kodakaraensis | Chitinase | 90 kDa | 70 °C, 3 h | (GlcNAc)2 | TLC | [52] |
| KOD1 | |||||||
| Chitin | Vibrio anguillarum E-383a | Exochitinase | - | - | (GlcNAc)2 40.3% | HPLC | [47] |
| Chitin | Enterobacter sp. G1 | Chitosanase | 50 kDa | 35 °C, 5 min | (GlcNAc)2 | TLC/HPLC | [53] |
| Chitosan (80% DD) | |||||||
| Chitin | Corynebacterium sp. | Chitobiase | - | 40 °C, 24 h | GlcNAc | HPLC/NMR | [54] |
| Chitin, steam exploded | Lecanicillium lecanii | Chitinase | 40 °C, 6 days | GlcNAcDP 1~9 | HPLC/MALDI-TOF MS | [55] | |
| (GlcNAc)2, | Stenotrophomonas maltophilia | N-acetyl-β-hexosaminidase & Chitin synthase | 40 °C, 60 min | GlcNAc | HPLC | [46] | |
| (GlcNAc)6 |
2.2. N,N′-Diacetylchitobiose and Chitin Oligosaccharides
3. Bioproduction of Chitosan Oligosaccharides and Its Monomer
| Chitosan Source | Enzyme Source | Enzyme | Mol. Wt. | Condition | Product & Yield | Analysis | Reference |
|---|---|---|---|---|---|---|---|
| Chitosan | Bacillus sp. KCTC 0377BP | Chitosanase | 45 kDa | 1700 (unit/mg) | (GlcN)3~(GlcN)7 | TLC/HPLC | [64] |
| Chitosan | Bacillus cereus S1 | Chitosanase | 45 kDa | 40 °C, 20 min | (GlcN)2 27.2% | HPLC | [65] |
| (GlcN)3 40.6% | |||||||
| (GlcN)4 32.2% | |||||||
| Chitosan | Bacillus sp. 16 | Chitosanase | 37 °C, 30 min | DP 2~9 (DP 5~6) | TLC/HPLC | [66] | |
| Chitosan | Bacillus sp. KFB-C108 | Chitosanase | 48 kDa | 55 °C, 12 h | (GlcN)3~(GlcN)5 | HPLC | [67] |
| Chitosan | Bacillus sp. HW-002 | Chitosanase | 46 kDa | 40 °C, 5 h | (GlcN)2 | HPLC | [68] |
| Chitosan | Bacillus pumilus BN-262 | Chitosanase | - | 45 °C, 1 h | (GlcN)4~(GlcN)6 | HPLC (NH2-60) Bio-Gel P-4gel | [69] |
| (GlcN)5~(GlcN)7 | |||||||
| Chitosan | Bacillus megaterium P1 | Chitosanase (A/B/C) | 43/39.5/22 kDa | 28 °C, 12 h/90 h | (GlcN)n oligomers | TLC | [70] |
| Chitosan | Acinetobacter sp. CHB101 | ChitosanaseI (endo) | 37 k Da | 37 °C, overnight | >(GlcN)5 | TLC | [71] |
| Chitosanase II (endo) | 30 kDa | ||||||
| Chitosan (60% DD) | Acinetobacter calcoaceticus TKU024 | Chitosanase (CHSA1) | 27 kDa | 37 °C, 30min | (GlcN)n oligomers | - | [72] |
| Chitosanase (CHSA2) | 66 kDa | ||||||
| Chitosan | Nocardia orientalis IFO 12806 | Chitosanase (exo) | 97 kDa (70 kDa) | 40 °C, 24 h | GlcN | TLC/HPLC | [73] |
| Chitosan | Matsuebacter chitosanotabidus 3001 | Chitosanase | 34 kDa | 30 °C, 10 min | (GlcN)2~(GlcN)6 | TLC | [74] |
| Chitosan | Aspergillus fumigatus KH-94 | Chitosanase I (endo) | 22.5 kDa | 50 °C, 30 min | DP 3~6 50% & >DP7 50% | TLC/HPLC | [75] |
| Chitosanase II (exo) | 108 kDa | 50 °C, 5 min | GlcN | ||||
| Chitosan | Aspergillus fumigates S-26 | Chitosanase | 104 kDa | 37 °C, 30 min | GlcN | TLC/HPLC | [76] |
| (GlcN)2~(GlcN)7 | |||||||
| Chitosan | Aspergillus oryzae IAM2660 | Chitosanase(endo) Chitosanase (exo) | 40 kDa | 37 °C, 20 min | >DP 5 | TLC | [77] |
| 135 kDa | 40 °C, overnight | GlcN | |||||
| Chitosan | Trichoderma reesei PC-3-7 | Chitosanase | 93 kDa | 37 °C, 15 h | GlcN | TLC | [78] |
| Chitosan | - | Immobilized papain | - | - | MW < 10,000 49.5% | MALDI-TOF MS | [79] |
| MW 600~2000 11.1% | |||||||
| Chitosan(82.8% DD) | Novozyme lipase | - | 37 °C, 24 h | DP 1–6 | TLC | [80] | |
| Chitosan (76% DD) | Commercial enzymes | Complex (cellulase, α-amylase, proteinase) | - | 40 °C, 40 min | DP 5~17 | MALDI-TOF-MS | [81] |
| Chitosan (95% DD) | Bacillus cereus TNU-FC-4 | Chitosanase | 46 kDa | 45 °C, 33 min | >DP 7 | HPLC | [25] |
| Rhizopus oligosporus cell wall | Streptomyces sp. N174 | Chitosanase | - | 40 °C, 24 h | (GlcN)2, GlcN-GlcNAc, (GlcN)2-GlcNAc | CP/MAS NMR/MALDI-TOF-MS | [ 82] |
| Chitosan | Rhodothermus obamensis | Branchzyme | 256 kDa | 50 °C, 24 h | DP 2–20 | GC-FID/SEC-HPLC | [83] |
| Chitosan (60% DD) | Penicillium janthinellum D4 | Chitosanase | 49 kDa | 50 °C, 60 h | DP 3–9 | MALDI-TOF-MS | [84] |
3.1. β-d-Glucosamine
3.2. Chitobiose and Chitosan Oligosaccharides
4. Chitin Deacetylase and Chitooligosaccharides
| Product | Reaction Type | Reference |
|---|---|---|
| (GlcN)2~(GlcN)6 | N-Deacetylation with endochitinase and CDA from Aspergillus nidulans; substrates: chitin | [97] |
| GlcN-GlcNAc | N-Deacetylation with CDA from C. lindemuthianum ATCC 56676; substrates: (GlcNAc)2 | [96] |
| (GlcN)3 & (GlcN)4 | (GlcNAc)3 and (GlcNAc)4 | |
| Chitosan oligomers | N-Deacetylation with CDA from Mucor rouxii; substrates: partially N-acetylated chitosans (DPn, n = 30) | [100] |
| GlcN | N-Deacetylation with CDA from Thermococcus kodakaraensisKOD1; substrates: GlcNAc | [95] |
| (GlcN)2~(GlcN)7 | N-Deacetylation with CDA from Absidia corymbifera DY-9; substrates: (GlcNAc)2~(GlcNAc)7 and WSCT-50 | [94] |
| (GlcN)2~(GlcN)7 | N-Deacetylation with CDA from Mortierella sp. DY-52; substrates: (GlcNAc)2~(GlcNAc)7 | [91] |
| Chitosan (89% DD) & (GlcN)2~(GlcN)4 | N-Deacetylation with CDA from Saccharomyces cerevisiae; substrates: Chitin and (GlcNAc)2~(GlcNAc)4 | [98] |
| (GlcNAc)6~(GlcNAc)15 | Transglycosylation reaction on β-1,4-(GlcNAc)3with lysozyme containing (NH4)2SO4 (30% w/v) | [101] |
| β-1,4-(GlcNAc)2 & β-1,6-(GlcNAc)2 | Transglycosylation reaction on N-acetylchito-oligosaccharides [β-1,4-(GlcNAc)2 ~ β-1,4-(GlcNAc)6] with exo-β-d-GlcNase from Alteromonas sp. OK2607 | [102] |
| n-Butyl β-d-glucosaminide (C4GlcN) | Transglycosylation reaction on chitosan oligosaccharides & n-butanol with exo-β-d-GlcNase from Penicillium funiculosum KY616 | [103] |
5. Transglycosylation and Chitooligosaccharides
6. Chemoenzymatic Glycosylation and Chitooligosaccharides
| Product | Reaction Type | Reference |
|---|---|---|
| N-Acetylchitooligosaccharides | Chemoenzymatic elongation of N-GlcNAc unit by combined use of chitinase and β-galactosidase | [115] |
| Chitin derivatives with the deacetylated extents ranging from 0% to 50% | Chitinase-catalyzed copolymerization of N-acetylchitobiose oxazoline with the N,N′-diacetylchitobiose oxazoline | [54] |
| 6- O-Carboxymethylated chitotetraose alternatingly | Chitinase-catalyzed chemoenzymatic glycosylation of 6- O- and 6′-O-carboxymethyled chitobioseoxazolines | [119] |
| 3- O-Methylated chitotetraose | Chitinase-catalyzed chemoenzymatic glycosylation with 3- O-methyl- and 3′-O-methylchitobiose oxazolines | [118] |
| Oligo- N-acetyllactosamine derivatives with β-(1→4)-β-(1→6)-linked repeating unit | Chemoenzymatic polymerization by using transition state analogue substrate with chitinase A1 | [116] |
| Alternatingly d-Glcβ-(1→4) N-GlcNAc repeating units, a cellulose-chitin hybrid polysaccharide | Chemoenzymatic glycosylation of Glcβ (1→4) GlcN Acoxazoline and GlcNAcβ (1→4) Glc fluoride by chitinase and cellulose, respectively | [103] |
| Alternatingly d-GlcNβ-(1→4) N-GlcNAc repeating units, a chitin-chitosan hybrid polysaccharide | Chitinase-catalyzed chemoenzymatic glycosylation of C-2′ position modified N-acetylchitobiose oxazolines | [55] |
| Fluorinated chitins | Chitinase-catalyzed polymerization C-6, C-6′, or both modified N-acetylchitobiose oxazolines | [120,121] |
7. Perspectives
7.1. Direct Degradation and Separation of Chitin from Crustacean Shells or Squid Pens
7.2. New Enzymes with Better Properties
7.3. Manipulation of the Size-Distribution of Oligomers Produced by Enzymatic Bioconversion
7.4. Genetic Engineering Technology
7.5. Direct Fermentation of Raw Materials such as Crab and Shrimp Shells
7.6. Reactor Systems
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- No, H.K.; Meyers, S.P.; Lee, K.S. Isolation and characterization of chitin from crawfish shell waste. J. Agric. Food Chem. 1989, 37, 575–579. [Google Scholar]
- Aye, K.N.; Stevens, W.F. Technical note: Improved chitin production by pre-treatment of shrimp shells. J. Chem. Technol. Biotechnol. 2004, 79, 421–425. [Google Scholar] [CrossRef]
- Allan, G.G.; Fox, J.R.; Kong, N. Marine polymers, part 8. A critical evaluation of the potential sources of chitin and chitosan. In Proceeding of the 1st International Conference on Chitin/Chitosan; Muzzarelli, R.A.A., Pariser, E.R., Eds.; MIT Sea Grant: Cambridge, MA, USA, 1978; pp. 64–78. [Google Scholar]
- Simpson, B.K.; Gagné, N.; Simpson, M.V. Bioprocessing of chitin and chitosan. In Fisheries Processing: Biotechnological Applications; Martin, A.M., Ed.; Chapman & Hall: London, UK, 1994; pp. 155–173. [Google Scholar]
- Healy, M.G.; Romo, C.R.; Bustos, R. Bioconversion of marine crustacean shell waste. Res. Conserv. Recycl. 1994, 11, 139–147. [Google Scholar] [CrossRef]
- Jung, W.J.; Jo, G.Y.; Kuk, J.H.; Kim, K.Y.; Park, R.D. Extraction of chitin from red crab shell waste by cofermentation with Lactobacillus paracasei subsp. tolerans KCTC-3074 and Serratia marcescens FS-3. Appl. Microbiol. Biot. 2006, 71, 234–237. [Google Scholar]
- Jung, W.J.; Jo, G.Y.; Kuk, J.H.; Kim, Y.J.; Oh, K.T.; Park, R.D. Production of chitin from red crab shell waste by successive fermentation with Lactobacillus paracasei KCTC-3074 and Serratia marcescens FS-3. Carbohydr. Polym. 2007, 68, 746–750. [Google Scholar] [CrossRef]
- Jo, G.H.; Jung, W.J.; Kuk, J.H.; Oh, K.T.; Kim, Y.J.; Park, R.D. Screening of protease-producing Serratia marcescens FS-3 and its application to deproteinization of crab shell waste for chitin extraction. Carbohydr. Polym. 2008, 74, 504–508. [Google Scholar] [CrossRef]
- Seyfarth, F.; Schliemann, S.; Elsner, P.; Hipler, U.C. Antifungal effect of high- and low-molecular-weight chitosan hydrochloride, carboxymethyl chitosan, chitosan oligosaccharide and N-acetyl-d-glucosamine against Candida albicans, Candida krusei and Candida glabrata. Int. J. Pharm. 2008, 353, 139–148. [Google Scholar] [PubMed]
- Oliveira, E.N., Jr.; El Gueddari, N.E.; Moerschbacher, B.M.; Peter, M.G.; Franco, T.T. Growth of phytopathogenic fungi in the presence of partially acetylated chitooligosaccharides. Mycopathologia 2008, 166, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, C.; Fukamizo, T. Production of food oligosaccharides by chitinases and chitosanases: Structural biological perspective. New Food Ind. 2001, 43, 7–14. [Google Scholar]
- Lee, H.W.; Park, Y.S.; Choi, J.W.; Yi, S.Y.; Shin, W.S. Antidiabetic effects of chitosan oligosaccharides in neonatal streptozotocin-induced noninsulin-dependent diabetes mellitus in rats. Biol. Pharm. Bull. 2003, 26, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Liu, P.; Zhang, J.; Chen, J. Biological activities of chitosan and chitooligosaccharides. Food Hydrocoll. 2011, 25, 170–179. [Google Scholar] [CrossRef]
- Sato, K.; Saimoto, H.; Morimoto, M.; Shigemasa, Y. Depolymerization of chitin and chitosan under hydrothermal conditions. Sen-I Gakkaishi 2003, 59, 104–109. [Google Scholar] [CrossRef]
- Xing, R.E.; Liu, S.; Yu, H.H.; Guo, Z.Y.; Wang, P.B.; Li, C.P.; Li, Z.; Li, P.C. Salt-assisted acid hydrolysis of chitosan to oligomers under microwave irradiation. Carbohydr. Res. 2005, 340, 2150–2153. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zivanovic, S.; Hayes, D.G.; Weiss, J. Efficient reduction of chitosan molecular weight by high-intensity ultrasound: Underlying mechanism and effect of process parameters. J. Agric. Food Chem. 2008, 56, 5112–5119. [Google Scholar] [CrossRef] [PubMed]
- Yoksan, R.; Akashi, M.; Miyata, M.; Chirachanchai, S. Optimal gamma-ray dose and irradiation conditions for producing low-molecular-weight chitosan that retains its chemical structure. Radiat. Res. 2004, 161, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Domard, A.; Cartier, N. Glucosamine oligomers: 4. Solid state-crystallization and sustained dissolution. Int. J. Biol. Macromol. 1992, 14, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Einbu, A.; Vårum, K.M. Depolymerization and de-N-acetylation of chitin oligomers in hydrochloric acid. Biomacromolecules 2007, 8, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Jia, X.G.; Lei, W.X.; Li, Z.J.; Zhang, T.Y. Spectra analyses of chitosans degraded by hydrogen peroxide under optimal conditions. Spectrosc. Spectr. Anal. 2009, 29, 43–47. [Google Scholar]
- Morris, V.B.; Neethu, S.; Abraham, T.E.; Pillai, C.K.S.; Sharma, C.P. Studies on the condensation of depolymerized chitosans with DNA for preparing chitosan-DNA nanoparticles for gene delivery applications. J. Biomed. Mater. Res. Part B 2009, 89B, 282–292. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Park, P.J.; Kim, S.K. Antimicrobial effect of chitooligosaccharides produced by bioreactor. Carbohydr. Polym. 2001, 44, 71–76. [Google Scholar]
- Qin, C.; Zhou, B.; Zeng, L.; Zhang, Z.; Liu, Y.; Du, Y.; Xiao, L. The physicochemical properties and antitumor activity of cellulase-treated chitosan. Food Chem. 2004, 84, 107–115. [Google Scholar]
- Aam, B.B.; Heggset, E.B.; Norberg, A.L.; Sørlie, M.; Vårum, K.M.; Eijsink, V.G.H. Production of chitooligosaccharides and their potential applications in medicine. Mar. Drugs 2010, 8, 1482–1517. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W.; Hsiao, Y.C.; Chiang, B.H. Production of high degree polymerized chitooligosaccharides in a membrane reactor using purified chitosanase from Bacillus cereus. Food Res. Int. 2009, 42, 1355–1361. [Google Scholar] [CrossRef]
- Zeng, H.; Zheng, L.Y. Studies on Penicillium sp. ZDZ1 chitosanase immobilized on chitin by cross-linking reaction. Process Biochem. 2002, 38, 531–535. [Google Scholar]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active enZYmes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- CAZy. The Carbohydrate-Active enZYmes database. Available online: http://www.cazy.org (accessed on 27 April 2010).
- Davies, G.; Henrissat, B. Structures and mechanisms of glycosyl hydrolases. Structure 1995, 3, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Henrissat, B.; Bairoch, A. Updating the sequence-based classification of glycosyl hydrolases. Biochem. J. 1996, 316, 695–696. [Google Scholar] [PubMed]
- Kobayashi, S.; Kiyosada, T.; Shoda, S. Synthesis of artificial chitin: Irreversible catalytic behavior of a glycosyl hydrolase through a transition state analogue substrate. J. Am. Chem. Soc. 1996, 118, 13113–13114. [Google Scholar]
- Sato, H.; Mizutani, S.; Tsuge, S.; Ohtani, H.; Aoi, K.; Takasu, A.; Okada, M.; Kobayashi, S.; Kiyosada, T.; Shoda, S. Determination of the degree of acetylation of chitin/chitosan by pyrolysis-gas chromatography in the presence of oxalic acid. Anal. Chem. 1998, 70, 7–12. [Google Scholar] [CrossRef]
- ExplorEnz—The Enzyme Database. Available online: http://www.enzyme-explorer.org/ (accessed on 27 April 2010).
- Berger, L.R.; Reynolds, D.M. The chitinase system of a strain of Streptomyces griseus. Biochem. Biophys. Acta 1958, 29, 522–534. [Google Scholar]
- Monreal, J.; Reese, E.T. The chitinase of Serratia marcescens. J. Microbiol. 1969, 15, 689–696. [Google Scholar]
- Sørbotten, A.; Horn, S.J.; Eijsink, V.G.; Vårum, K.M. Degradation of chitosans with chitinase B from Serratia marcescens. Production of chito-oligosaccharides and insight into enzyme processivity. FEBS J. 2005, 272, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Lopatin, S.A.; Derbeneva, M.S.; Kulikov, S.N.; Varlamov, V.P.; Shpigun, O.A. Fractionation of chitosan by ultrafiltration. J. Anal. Chem. 2009, 64, 648–651. [Google Scholar] [CrossRef]
- Haebel, S.; Bahrke, S.; Peter, M.G. Quantitative sequencing of complex mixtures of heterochitooligosaccharides by MALDI-linear ion trap mass spectrometry. Anal. Chem. 2007, 79, 5557–5566. [Google Scholar]
- Le Dévédec, F.; Bazinet, L.; Furtos, A.; Venne, K.; Brunet, S.; Mateescu, M.A. Separation of chitosan oligomers by immobilized metal affinity chromatography. J. Chromatogr. A 2008, 1194, 165–171. [Google Scholar] [CrossRef]
- Pichyangkura, R.; Kudan, S.; Kuttiyawong, K.; Sukwattanasinitt, M.; Aiba, S. Quantitative production of 2-acetamido-2-deoxy-d-glucose from crystalline chitin by bacterial chitinase. Carbohydr. Res. 2002, 337, 557–559. [Google Scholar] [CrossRef]
- Sashiwa, H.; Fujishima, S.; Yamano, N.; Kawasaki, N.; Nakayama, A.; Muraki, E.; Sukwattanasinitt, M.; Pichyangkurac, R.; Aiba, S. Enzymatic production of N-acetyl-d-glucosamine from chitin. Degradation study of N-acetylchitooligosaccharide and the effect of mixing of crude enzymes. Carbohydr. Polym. 2003, 51, 391–395. [Google Scholar] [CrossRef]
- Il’ina, A.V.; Zueva, O.Y.; Lopatin, S.A.; Varlamov, V.P. Enzymatic hydrolysis of α-chitin. Appl. Biochem. Microbiol. 2004, 40, 35–38. [Google Scholar]
- Sukwattanasinitt, M.; Zhu, H.; Sashiwa, H.; Aiba, S. Utilization of commercial non-chitinase enzymes from fungi for preparation of 2-acetamido-2-deox-d-glucose from β-chitin. Carbohydr. Res. 2002, 337, 133–137. [Google Scholar] [CrossRef]
- Sashiwa, H.; Fujishima, S.; Yamano, N.; Kawasaki, N.; Nakayama, A.; Muraki, E.; Hiraga, K.; Oda, K.; Aiba, S. Production of N-acetyl-d-glucosamine from α-chitin by crude enzymes from Aeromonas hydrophila H-2330. Carbohydr. Res. 2002, 337, 761–763. [Google Scholar] [CrossRef]
- Thamthiankul, S.; Suan-Ngay, S.; Tantimavanich, S.; Panbangred, W. Chitinase from Bacillus thuringiensis subsp pakistani. Appl. Microbiol. Biotechnol. 2001, 56, 395–401. [Google Scholar]
- Katta, S.; Ankati, S.; Podile, A.R. Chitooligosaccharides are converted to N-acetylglucosamine by N-acetyl-β-hexosaminidase from Stenotrophomonas maltophilia. FEMS Microbiol. Lett. 2013, 348, 19–25. [Google Scholar] [CrossRef]
- Lan, X.; Ozawa, N.; Nishiwaki, N.; Kodaira, R.; Okazaki, M.; Shimosaka, M. Purification, cloning, and sequence analysis of β-N-acetylglucosaminidase from the chitinolytic bacterium Aeromonas hydrophila strain SUWA-9. Biosci. Biotechnol. Biochem. 2004, 68, 1082–1090. [Google Scholar]
- Kuk, J.H.; Jung, W.J.; Jo, G.H.; Ahn, J.S.; Kim, K.Y.; Park, R.D. Selective preparation of N-acetyl-d-glucosamine and N,N′-diacetylchitobiose from chitin using a crude enzyme preparation from Aeromonas sp. Biotechnol. Lett. 2005, 27, 7–11. [Google Scholar] [CrossRef]
- Kuk, J.H.; Jung, W.J.; Jo, G.H.; Kim, Y.C.; Kim, K.Y.; Park, R.D. Production of N-acetyl-β-d-glucosamine from chitin by Aeromonas sp. GJ-18 crude enzyme. Appl. Microbiol. Biotechnol. 2005, 68, 384–389. [Google Scholar] [CrossRef]
- Kuk, J.H.; Jung, W.J.; Jo, G.H.; Kim, K.Y.; Park, R.D. Production of N,N′-diacetylchitobiose from chitin using temperature-sensitive chitinolytic enzyme preparations of Aeromonas sp. GJ-18. World J. Microbiol. Biotechnol. 2006, 22, 135–139. [Google Scholar] [CrossRef]
- Martinez, E.A.; Boer, H.; Koivula, A.; Samain, E.; Driguez, H.; Armand, S.; Cottaz, S. Engineering chitinases for the synthesis of chitin oligosaccharides: Catalytic amino acid mutations convert the GH-18 family glycoside hydrolases into transglycosylases. J. Mol. Catalys. B Enzym. 2012, 74, 89–96. [Google Scholar]
- Tanaka, T.; Fukui, T.; Atomi, H.; Imanaka, T. Characterization of an exo-β-d-glucosaminidase involved in a novel chitinolytic pathway from the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Bact. 2003, 185, 5175–5181. [Google Scholar]
- Yamasaki, Y.; Hayashi, I.; Ohta, Y.; Nakagawa, T.; Kawamukai, M.; Matsuda, H. Purification and mode of action of chitosanolytic enzyme from Enterobacter sp. G-1. Biosci. Biotechnol. Biochem. 1993, 57, 444–449. [Google Scholar]
- Makino, A.; Sakamoto, J.; Ohmae, M.; Kobayashi, S. Effect of fluorine substituent on the chitinase-catalyzed polymerization of sugar oxazoline derivatives. Chem. Lett. 2006, 35, 160–161. [Google Scholar] [CrossRef]
- Makino, A.; Ohmae, M.; Kobayashi, S. Chitinase-catalyzed copolymerization to a chitin derivative having glucosamine unit in controlled proportion. Polym. J. 2006, 38, 1182–1188. [Google Scholar] [CrossRef]
- Takiguchi, Y.; Shimahara, K. N,N-Diacetylchitobiose production from chitin by Vibrio anguillarum strain E-383a. Lett. Appl. Microbiol. 1988, 6, 129–131. [Google Scholar] [CrossRef]
- Ilankovan, P.; Hein, S.; Ng, C.H.; Trung, T.S.; Stevens, W.F. Production of N-acetyl chitobiose from various chitin substrates using commercial enzymes. Carbohydr. Polym. 2006, 63, 245–250. [Google Scholar] [CrossRef]
- Wang, S.L.; Liu, C.P.; Liang, T.W. Fermented and enzymatic production of chitin/chitosan oligosaccharides by extracellular chitinases from Bacillus cereus TKU027. Carbohydr. Polym. 2012, 90, 1305–1313. [Google Scholar]
- Wang, C.Y.; Hsieh, Y.Z. Analysis of chitin oligosaccharides by capillary electrophoresis with laser-inducedfluorescence. J. Chromatogr. A 2002, 979, 431–438. [Google Scholar]
- Saito, J.; Kita, A.; Higuchi, Y.; Nagata, Y.; Ando, A.; Miki, K. Crystal structure of chitosanase from Bacillus circulans MH-K1 at 1.6-angstrom resolution and its substrate recognition mechanism. J. Biol. Chem. 1999, 274, 30818–30825. [Google Scholar] [CrossRef] [PubMed]
- Fukamizo, T.; Ohkawa, T.; Ikeda, Y.; Goto, S. Specificity of chitosanase from Bacillus pumilus. Biochim. Biophys. Acta 1994, 1205, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Chang, C.H.; Wu, Y.J.; Li, Y.K. Exploration of glycosyl hydrolase family 75, a chitosanase from Aspergillus fumigatus. J. Biol. Chem. 2006, 281, 3137–3144. [Google Scholar] [CrossRef]
- Fukamizo, T.; Brzezinski, R. Chitosanase from Streptomyces sp. strain N174: A comparative review of its structure and function. Biochem. Cell Biol. 1997, 75, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Kim, E.J.; Piao, Z.; Yun, Y.C.; Shin, Y.C. Purification and characterization of chitosanase from Bacillus sp. strain KCTC 0377BP and its application for the production of chitosan oligosaccharides. Appl. Environ. Microbiol. 2004, 70, 4522–4531. [Google Scholar]
- Kurakake, M.; You, S.; Nakagawa, K.; Sugihara, M.; Komaki, T. Properties of chitosanase from Bacillus cereus S1. Curr. Microbiol. 2000, 40, 6–9. [Google Scholar] [CrossRef]
- Park, R.D.; Rhee, C.O.; Lee, H.C.; Cho, C.S.; Jo, D.H. A comparative study of chitooligosaccharide production from chitosan. J. Chitin Chitosan 1999, 4, 96–101. [Google Scholar]
- Yoon, H.G.; Ha, S.C.; Lim, Y.H.; Cho, H.Y. New thermostable chitosanase from Bacillus sp.: Purification and characterization. J. Microbiol. Biotechnol. 1998, 8, 449–454. [Google Scholar]
- Lee, H.W.; Choi, J.W.; Han, D.P.; Park, M.J.; Lee, N.W.; Yi, D.H. Purification and characteristics of chitosanase from Bacillus sp. HW-002. J. Microbiol. Biotechnol. 1996, 6, 19–25. [Google Scholar]
- Jeon, Y.J.; Kim, S.K. Production of chitooligosaccharides using ultrafilteration membrane reactor and their antibacterial activity. Carbohydr. Polym. 2000, 41, 133–144. [Google Scholar] [CrossRef]
- Pelletier, A.; Sygusch, J. Purification and characterization of three chitosanase activities from Bacillus megaterium P1. Appl. Environ. Microbiol. 1990, 56, 844–848. [Google Scholar]
- Shimosaka, M.; Nogawa, M.; Wang, X.Y.; Kumehara, M.; Okazaki, M. Production of two chitosanases from a chitosan-assimilating bacterium, Acinetobacter sp. strain CHB101. Appl. Environ. Microbiol. 1995, 61, 438–442. [Google Scholar]
- Wang, S.L.; Tseng, W.N.; Liang, T.W. Biodegradation of shellfish wastes and production of chitosanases by a squid pen-assimilating bacterium, Acinetobacter calcoaceticus TKU024. Biodegradation 2011, 22, 939–948. [Google Scholar] [CrossRef]
- Nanjo, F.; Katsumi, R.; Sakai, K. Purification and characterization of an exo-β-d-glucosaminidase, a novel type of enzyme, from Nocardia orientalis. J. Biol. Chem. 1990, 265, 10088–10094. [Google Scholar]
- Park, J.K.; Shimono, K.; Ochiai, N.; Shigeru, K.; Kurita, M.; Ohta, Y.; Tanaka, K.; Matsuda, H.; Kawamukai, M. Purification, characterization, and gene analysis of a chitosanase (ChoA) from Matsuebacter chitosanotabidus 3001. J. Bact. 1999, 181, 6642–6649. [Google Scholar]
- Kim, S.Y.; Shon, D.H.; Lee, K.H. Purification and characteristics of two types of chitosanases from Aspergillus fumigatus. J. Microbiol. Biotechnol. 1998, 8, 568–574. [Google Scholar]
- Jung, W.J.; Kuk, J.H.; Kim, K.Y.; Jung, K.C.; Park, R.D. Purification and characterization of exo-β-d-glucosaminidase from Aspergillus fumigatus S-26. Protein Expr. Purif. 2006, 45, 125–131. [Google Scholar]
- Zhang, X.Y.; Dae, A.L.; Zhang, X.K.; Kuroiwa, K. Purification and characterization of chitosanase and exo-β-d-glucosaminidase from Koji Mold, Aspergillus oryzae IAM2660. Biosci. Biotechnol. Biochem. 2000, 64, 1896–1902. [Google Scholar]
- Nogawa, M.; Takahashi, H.; Kasiwagi, A.; Ohshima, K.; Okada, H.; Morikawa, Y. Purification and characterization of exo-β-d-glucosaminidase from a cellulolytic fungus, Trichoderma reesei PC-3-7. Appl. Envoron. Microbiol. 1998, 64, 890–895. [Google Scholar]
- Lin, H.; Wang, H.; Xue, C.; Ye, M. Preparation of chitosan oligomers by immobilized papain. Enzyme Microb. Technol. 2002, 31, 588–592. [Google Scholar] [CrossRef]
- Lee, D.X.; Xia, W.S.; Zhang, J.L. Enzymatic preparation of chitooligosaccharides by commercial lipase. Food Chem. 2008, 111, 291–295. [Google Scholar] [CrossRef]
- Zhang, H.; Du, Y.; Yu, X.; Mitsutomi, M.; Aiba, S. Preparation of chitooligosaccharides from chitosan by acomplex enzyme. Carbohydr. Res. 1999, 320, 257–260. [Google Scholar] [CrossRef]
- Mahata, M.; Shinya, S.; Masaki, E.; Yamamoto, T.; Ohnuma, T.; Brzezinski, R.; Mazumder, T.K.; Yamashita, K.; Narihiro, K.; Fukamizo, T. Production of chitooligosaccharides from Rhizopus oligosporus NRRL2710 cells by chitosanase digestion. Carbohydr. Res. 2014, 383, 27–33. [Google Scholar] [CrossRef]
- Yalpani, M.; Pantaleone, D. An examination of the unusual susceptibility of aminoglycans to enzymatic hydrolysis. Carbohydr. Res. 1994, 256, 159–175. [Google Scholar] [CrossRef]
- Nguyen, A.D.; Huang, C.C.; Liang, T.W.; Nguyen, V.B.; Pan, P.S.; Wang, S.L. Production and purification of a fungal chitosanase and chitooligomers from Penicillium janthinellum D4 and discovery of the enzyme activators. Carbohydr. Polym. 2014, 108, 331–337. [Google Scholar] [CrossRef]
- Gao, X.A.; Jung, W.J.; Kuk, J.H.; Park, R.D. Reaction pattern of Bacillus cereus D-11 chitosanase on chito-oligosaccharide alcohols. J. Microbiol. Biotechnol. 2009, 19, 358–361. [Google Scholar]
- Pantaleone, D.; Yalpani, M.; Scollar, M. Unusual susceptibility of chitosan to enzymic hydrolysis. Carbohydr. Res.1992, 1992, 237, 325–332. [Google Scholar]
- Aiba, S.I. Preparation of N-acetylchitooligosaccharides from lysozymatic hydroxylate of partially N-acetylated chitosans. Carbohydr. Res. 1994, 261, 297–306. [Google Scholar] [CrossRef]
- Kumar, A.B.V.; Gowda, L.R.; Tharanathan, R.N. Non-specific depolymerization of chitosan by pronase and characterization of the resultant products. Eur. J. Biochem. 2004, 271, 713–723. [Google Scholar] [CrossRef]
- Xie, Y.; Hu, J.; Wei, Y.; Hong, X. Preparation of chitooligosaccharides by the enzymatic hydrolysis of chitosan. Polym. Degrad. Stab. 2009, 94, 1895–1899. [Google Scholar] [CrossRef]
- Wu, S. Preparation of chitooligosaccharides from Clanis bilineata larvae skin and their antibacterial activity. Int. J. Biol. Macromol. 2012, 51, 1147–1150. [Google Scholar]
- Zhao, Y.; Park, R.D.; Muzzarelli, R.A.A. Chitin deacetylases: Properties and applications. Mar. Drugs 2010, 8, 24–46. [Google Scholar] [CrossRef]
- Zhao, Y.; Kim, Y.J.; Oh, K.T.; Nguyen, V.N.; Park, R.D. Production and characterization of extracellular chitin deacetylase from Absidia corymbifera DY-9. J. Korean Soc. Appl. Biol. Chem. 2010, 53, 119–126. [Google Scholar] [CrossRef]
- Zhao, Y.; Ju, W.T.; Jo, G.H.; Jung, W.J.; Park, R.D. Perspectives of chitin deacetylase research. In Biotechnology of Biopolymers; Elnashar, M., Ed.; InTech: Rijeka, Croatia, 2011; pp. 131–144. [Google Scholar]
- Kim, Y.J.; Zhao, Y.; Oh, K.T.; Nguyen, V.N.; Park, R.D. Enzymatic deacetylation of chitin by extracellular chitin deacetylase from newly screened Mortierella sp. DY-52. J. Microbiol. Biotechnol. 2008, 18, 759–766. [Google Scholar]
- Tanaka, T.; Fukui, T.; Fujiwara, S.; Atomi, H.; Imanaka, T. Concerted action of diacetylchitobiose deacetylase and exo-beta-d-glucosaminidase in a novel Biotechnology of Biopolymers chitinolytic pathway in the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Biol. Chem. 2004, 279, 30021–30027. [Google Scholar]
- Tokuyasu, K.; Ono, H.; Ohnishi-Kameyama, M.; Hayashi, K.; Mori, Y. Deacetylation of chitin oligosaccharides of dp 2-4 by chitin deacetylase from Colletotrichum lindemuthianum. Carbohydr. Res. 1997, 303, 353–358. [Google Scholar] [CrossRef]
- Alfonso, C.; Nuero, O.M.; Santamarla, F.; Reyes, F. Purification of a heat-stable chitin deacetylase from Aspergillus nidulans and its role in cell wall degradation. Curr. Microbiol. 1995, 30, 49–54. [Google Scholar] [CrossRef]
- Aguila, E.M.D.; Gomes, L.P.; Andrade, C.T.; Silva, J.T.; Paschoalin, V.M.F. Biocatalytic production of chitosan polymers from shrimp shells, using a recombinant enzyme produced by Pichia pastoris. Am. J. Mol. Biol. 2012, 2, 341–350. [Google Scholar] [CrossRef]
- Blair, D.E.; Hekmat, O.; Schuttelkopf, A.W.; Shrestha, B.; Tokuyasu, K.; Withers, S.G.; van Aalten, D.M. Structure and mechanism of chitin deacetylase from the fungal pathogen Colletotrichum lindemuthianum. Biochemistry 2006, 45, 9416–9426. [Google Scholar] [CrossRef] [PubMed]
- Martinou, A.; Bouriotis, V.; Stokke, B.T.; Vårum, K.M. Mode of action of chitin deacetylase from Mucor rouxii on partially N-acetylated chitosans. Carbohydr. Res. 1998, 311, 71–78. [Google Scholar] [CrossRef]
- Hattori, T.; Sakabe, Y.; Ogata, M.; Michishita, K.; Dohra, H.; Kawagishi, H.; Totani, K.; Nikaido, M.; Nakamura, T.; Koshino, H.; Usui, T. Enzymatic synthesis of an α-chitin-like substance via lysozyme-mediated transglycosylation. Carbohydr. Res. 2012, 347, 16–22. [Google Scholar] [CrossRef]
- Shimoda, K.; Nakajima, K.; Hiratsuka, Y.; Nishimura, S.I.; Kurita, K. Efficient preparation of β-(1→6)-(GlcNAc)2 by enzymatic conversion of chitin and chito-oligosaccharides. Carbohydr. Polym. 1996, 29, 149–154. [Google Scholar] [CrossRef]
- Matsumura, S.; Yao, E.; Toshima, K. One-step preparation of alkyl β-d-glucosaminide by the transglycosylation of chitosan and alcohol using purified exo-β-d-glucosaminidase. Biotechnol. Lett. 1999, 21, 451–456. [Google Scholar]
- Usui, T.; Matsu, H.; Isobe, K. Enzymic synthesis of useful chito-oligosaccharides utilizing transglycosylation by chitinolytic enzymes in a buffer containing ammonium sulfate. Carbohydr. Res. 1990, 203, 65–77. [Google Scholar] [CrossRef]
- Akiyama, K.; Kawazu, K.; Kobayashi, A. A novel method for chemo-enzymatic synthesis of elicitor-active chitosan oligomers and partially N-acetylated chitin oligomers using N-acetylated chitotriose as substrates in a lysozyme-catalyzed transglycosylation reaction system. Carbohydr. Res. 1995, 279, 151–160. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Hattori, T.; Uno, S.; Murata, T.; Usui, T. Enzymatic synthesis of gentiooligosaccharides by transglycosylation with β-glycosidases from Penicillium multicolor. Carbohydr. Res. 2009, 344, 972–978. [Google Scholar] [CrossRef]
- Kobayashi, S.; Makino, A.; Matsumoto, H.; Kunii, S.; Ohmae, M.; Kiyosada, T.; Makiguchi, K.; Matsumoto, A.; Horie, M.; Shoda, S.I. Enzymatic polymerization to novel polysaccharides having a glucose-N-acetylglucosamine repeating unit, a cellulose-chitin hybrid polysaccharide. Biomacromolecules 2006, 7, 1644–1656. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Makino, A. Enzymatic polymer synthesis: An opportunity for green polymer chemistry. Chem. Rev. 2009, 109, 5288–5353. [Google Scholar] [CrossRef]
- Kadokawa, J.I. Precision polysaccharide synthesis catalyzed by enzymes. Chem. Rev. 2011, 111, 4308–4345. [Google Scholar]
- Shoda, S.; Izumi, R.; Fujita, M. Green process in glycotechnology. Bull. Chem. Soc. Jpn. 2003, 76, 1–13. [Google Scholar]
- Kadokawa, J.; Shoda, S.J. New methods for architectures of glyco-materials. Synthetic Org. Chem. Jpn. 2003, 61, 1207. [Google Scholar] [CrossRef]
- Misawa, Y.; Lohavisavapanichi, C.; Shoda, S. Chemo-enzymatic synthesis of oligosaccharides having a polymerizable group at the reducing end. Glycoconj. J. 1999, 16, S122. [Google Scholar]
- Shoda, S.; Fujita, M.; Lohavisavapanichi, C.; Misawa, Y.; Ushizaki, K.; Tawata, Y.; Kuriyama, M.; Kohri, M.; Kuwata, H.; Watanabe, T. Efficient method for the elongation of the N-acetylglucosamine unit by combined use of chitinase and β-galactosidase. Helv. Chim. Acta 2002, 85, 3919. [Google Scholar] [CrossRef]
- Kadokawa, J.; Shoda, S. Enzymatic synthesis of glyco-macromonomers. Sen’i Gakkaishi 2003, 59, 74–78. [Google Scholar] [CrossRef]
- Shoda, S.; Misawa, Y.; Nishijima, Y.; Tawata, Y.; Kotake, T.; Noguchi, M.; Kobayashi, A.; Watanabe, T. Chemo-enzymatic synthesis of novel oligo-N-acetyllactosamine derivatives having a β(1-4)–β(1-6) repeating unit by using transition state analogue substrate. Cellulose 2006, 13, 477–484. [Google Scholar] [CrossRef]
- Watanabe, T.; Kobori, K.; Miyashita, K.; Fujii, T.; Sakai, H.; Uchida, M.; Tanaka, H. Identification of glutamic acid 204 and aspartic acid 200 in chitinase A1 of Bacillus circulans WL-12 as essential residues for chitinase activity. J. Biol. Chem. 1993, 268, 18567–18572. [Google Scholar]
- Watanabe, T.; Suzuki, K.; Oyanagi, W.; Ohnishi, K.; Tanaka, H. Gene cloning of chitinase A1 from Bacillus circulans WL-12 revealed its evolutionary relationship to Serratia chitinase and to the type III homology units of fibronectin. J. Biol. Chem. 1990, 265, 15659–15665. [Google Scholar]
- Sakamoto, J.; Watanabe, T.; Ariga, Y.; Kobayashi, S. Ring-opening glycosylation of a chitobiose oxazoline catalyzed by a non-chitinolytic mutant of chitinase. Chem. Lett. 2001, 30, 1180–1181. [Google Scholar]
- Ochiai, H.; Ohmae, M.; Kobayashi, S. Enzymatic synthesis of alternatingly 6-O-carboxymethylated chitotetraose by selective glycosidation with chitinase catalysis. Chem. Lett. 2004, 33, 694–695. [Google Scholar] [CrossRef]
- Ochiai, H.; Ohmae, M.; Kobayashi, S. Enzymatic glycosidation of sugar oxazolines having a carboxylate group catalyzed by chitinase. Carbohydr. Res. 2004, 339, 2769–2788. [Google Scholar] [CrossRef]
- Makino, A.; Kurosaki, K.; Ohmae, M.; Kobayashi, S. Chitinase-catalyzed synthesis of alternatingly N-Deacetylated chitin: A chitin–chitosan hybrid polysaccharide. Biomacromolecules 2006, 7, 950–957. [Google Scholar] [CrossRef]
- Villa-Lerma, G.; Gonzalez-Marquez, H.; Gimeno, M.; Lopez-Luna, A.; Barzana, E.; Shirai, K. Ultrasonication and steam-explosion as chitin pretreatments for chitin oligosaccharide production by chitinases of Lecanicillium lecanii. Biores. Technol. 2013, 146, 794–798. [Google Scholar]
- Nakagawa, Y.S.; Oyama, Y.; Kon, N.; Nikaido, M.; Tanno, K.; Kogawa, J.; Inomata, S.; Masui, A.; Yamamura, A.; Kawaguchi, M.; et al. Development of innovative technologies to decrease the environmental burdens associated with using chitin as a biomass resource: Mechanochemical grinding and enzymatic degradation. Carbohydr. Polym. 2011, 83, 1843–1849. [Google Scholar] [CrossRef]
- Sato, T.; Asada, K.; Takeda, M.; Tanno, K. Mechano-chemical synthesis of compound powders in ZnO–TiO2 system by a new high intensive ball mill. J. Jpn. Soc. Powder Metall. 2006, 53, 62–67. [Google Scholar]
- Takeda, T.; Nikaido, M.; Totani, T.; Obara, M.; Nakano, Y.; Uchimiya, H. Evaluation of high intensive ball mill method for effective saccharification of plant cell wall materials. J. Appl. Glycosci. 2009, 56, 71–76. [Google Scholar]
- Wu, Q.; Miao, Y. Mechanochemical effects of micronization on enzymatic hydrolysis of corn flour. Carbohydr. Polym. 2008, 72, 398–402. [Google Scholar]
- Fujimoto, S.; Inoue, H.; Yano, S.; Sakaki, T.; Minowa, T.; Endo, T. Bioethanol production from lignocellulosic biomass requiring no sulfuric acid: Mechanochemical pretreatment and enzymic saccharification. J. Jpn. Petrol. Inst. 2008, 51, 264–273. [Google Scholar] [CrossRef]
- Van Craeyveld, V.; Delcour, J.A.; Courtin, C.M. Ball milling improves extractability and affects molecular properties of Psyllium (Plantago ovate Forsk) seed husk arabinoxylan. J. Agric. Food Chem. 2008, 56, 11306–11311. [Google Scholar] [CrossRef]
- Reese, E.T.; Siu, R.G.H.; Levinson, H.S. The biological degradation of soluble cellulose derivatives and its relationship to the mechanism of cellulose hydrolysis. J. Bacteriol. 1950, 59, 485–497. [Google Scholar]
- Vaaje-Kolstad, G.; Westereng, B.; Horn, S.J.; Liu, Z.; Zhai, H.; Sørlie, M.; Eijsink, V.G.H. An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 2010, 330, 219–222. [Google Scholar] [CrossRef]
- Shinohara, M.L.; Ihara, M.; Abo, M.; Hashida, M.; Takagi, S.; Beck, T.C. A novel thermostable branching enzyme from an extremely thermophilic bacterial species, Rhodothermus obamensis. Appl. Microbiol. Biotechnol. 2001, 57, 653–659. [Google Scholar]
- Montilla, A.; Ruiz-Matute, A.I.; Corzo, N.; Cecilia Giacomini, C.; Irazoqui, G. Enzymatic generation of chitooligosaccharides from chitosan using soluble and immobilized glycosyltransferase (Branchzyme). J. Agric. Food Chem. 2013, 61, 10360–10367. [Google Scholar]
- Heggset, E.B.; Dybvik, A.I.; Hoell, I.A.; Norberg, A.L.; Sørlie, M.; Eijsink, V.G.H.; Vårum, K.M. Degradation of chitosans with a family 46 chitosanase from Streptomyces coelicolor A3(2). Biomacromolecules 2010, 11, 2487–2497. [Google Scholar] [CrossRef]
- Horn, S.J.; Sørbotten, A.; Synstad, B.; Sikorski, P.; Sørlie, M.; Vårum, K.M.; Eijsink, V.G. Endo/exo mechanism and processivity of family 18 chitinases produced by Serratia marcescens. FEBS J. 2006, 273, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Fukamizo, T.; Honda, Y.; Goto, S.; Boucher, I.; Brzezinski, R. Reaction mechanism of chitosanase from Streptomyces sp. N174. Biochem. J. 1995, 311, 377–383. [Google Scholar]
- Amano, K.; Ito, E. The action of lysozyme on partially deacetylated chitin. Eur. J. Biochem. 1978, 85, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, P.; Stokke, B.T.; Sørbotten, A.; Vårum, K.M.; Horn, S.J.; Eijsink, V.G. Development and application of a model for chitosan hydrolysis by a family 18 chitinase. Biopolymers 2005, 77, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Heggset, E.B.; Hoell, I.A.; Kristoffersen, M.; Eijsink, V.G.; Vårum, K.M. Degradation of chitosans with chitinase G from Streptomyces coelicolor A3(2): Production of chito-oligosaccharides and insight into subsite specificities. Biomacromolecules 2009, 10, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Mitsutomi, M.; Hata, T.; Kuwahara, T. Purification and characterization of novel chitinases from Streptomyces griseus Hut 6037. J. Ferment. Bioeng. 1995, 80, 153–158. [Google Scholar] [CrossRef]
- Sasaki, C.; Vårum, K.M.; Itoh, Y.; Tamoi, M.; Fukamizo, T. Rice chitinases: Sugar recognition specificities of the individual subsites. Glycobiology 2006, 16, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.J.; Sikorski, P.; Cederkvist, J.B.; Vaaje-Kolstad, G.; Sørlie, M.; Synstad, B.; Vriend, G.; Vårum, K.M.; Eijsink, V.G.H. Costs and benefits of processivity in enzymatic degradation of recalcitrant polysaccharides. Proc. Natl. Acad. Sci. USA 2006, 103, 18089–18094. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, P.; Sørbotten, A.; Horn, S.J.; Eijsink, V.G.; Vårum, K.M. Serratia marcescens chitinases with tunnel-shaped substrate-binding grooves show endo activity and different degrees of processivity during enzymatic hydrolysis of chitosan. Biochemistry 2006, 45, 9566–9574. [Google Scholar] [CrossRef] [PubMed]
- Gyore, J.; Parameswar, A.R.; Hebbard, C.F.F.; Oh, Y.; Bi, E.; Demchenko, A.V.; Price, N.P.; Orlean, P. 2-Acylamido analogues of N-acetylglucosamine prime formation of chitin oligosaccharides by yeast chitin synthase 2. J. Biol. Chem. 2014, 289, 12835–12841. [Google Scholar] [CrossRef]
- Liang, T.W.; Hsieh, J.L.; Wang, S.L. Production and purification of a protease, a chitosanase, and chitin oligosaccharides by Bacillus cereus TKU022 fermentation. Carbohydr. Res. 2012, 362, 38–46. [Google Scholar] [CrossRef]
- Vandanarachchi, J.K.; Kurukulasuriya, M.S.; Kim, S.K. Chitin, chitosan, and their oligosaccharides in food industry. In Chitin, Chitosan, Oligosaccharides and Their Derivatives; Kim, S.K., Ed.; CRC Press: Boca Raton, FL, USA, 2011; pp. 543–560. [Google Scholar]
- Jeon, Y.J.; Kim, S.K. Continuous production of chitooligosaccharides using a dual reactor system. Process Biochem. 2000, 35, 623–632. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, W.-J.; Park, R.-D. Bioproduction of Chitooligosaccharides: Present and Perspectives. Mar. Drugs 2014, 12, 5328-5356. https://doi.org/10.3390/md12115328
Jung W-J, Park R-D. Bioproduction of Chitooligosaccharides: Present and Perspectives. Marine Drugs. 2014; 12(11):5328-5356. https://doi.org/10.3390/md12115328
Chicago/Turabian StyleJung, Woo-Jin, and Ro-Dong Park. 2014. "Bioproduction of Chitooligosaccharides: Present and Perspectives" Marine Drugs 12, no. 11: 5328-5356. https://doi.org/10.3390/md12115328