Cell Permeable Peptides: A Promising Tool to Deliver Neuroprotective Agents in the Brain

{kind=link}
{kind=link}
Abstract
:1. Introduction
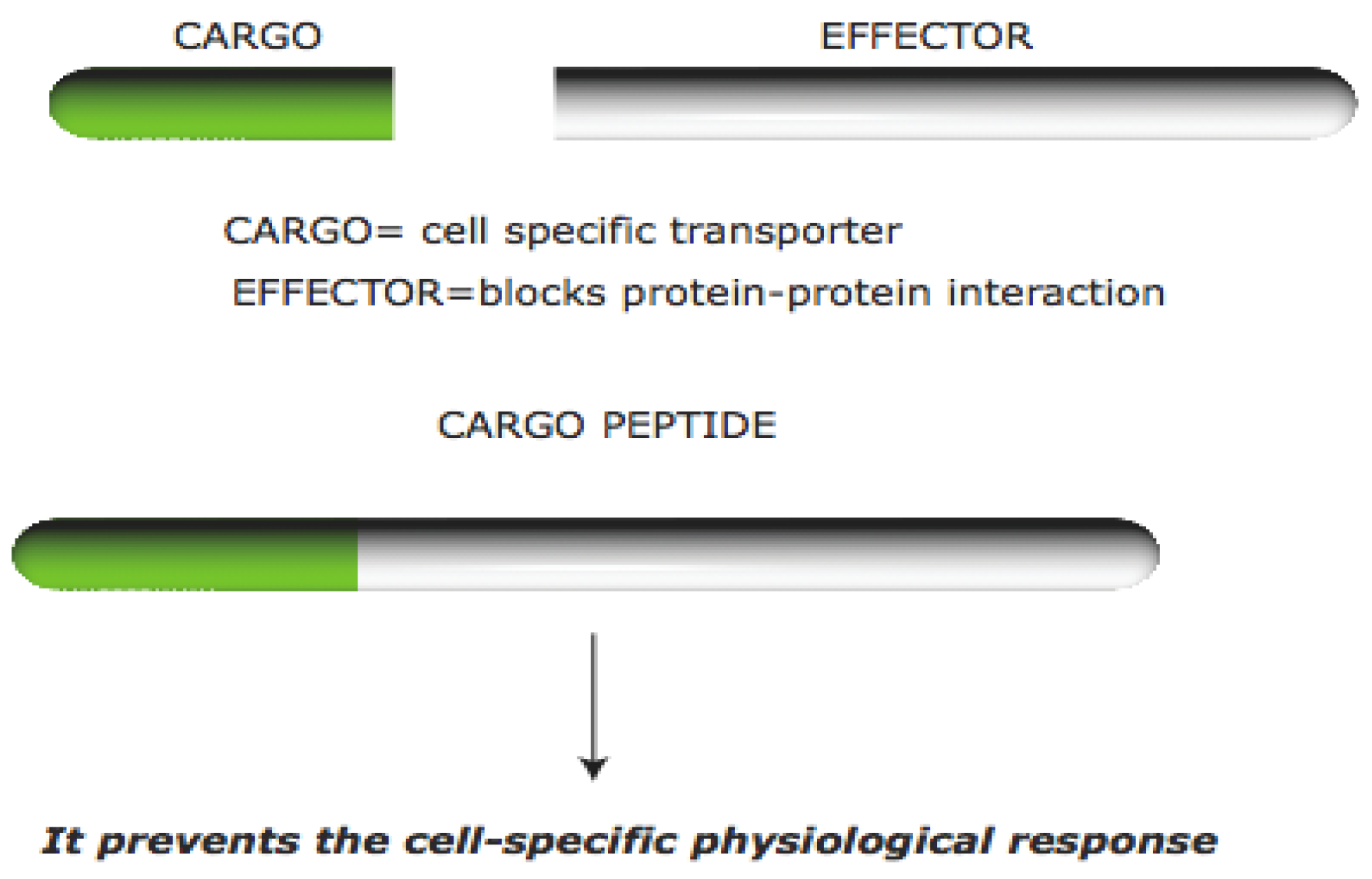
1.1. Cell Permeable Peptides

1.2. Protein–Protein Interactions in the Brain
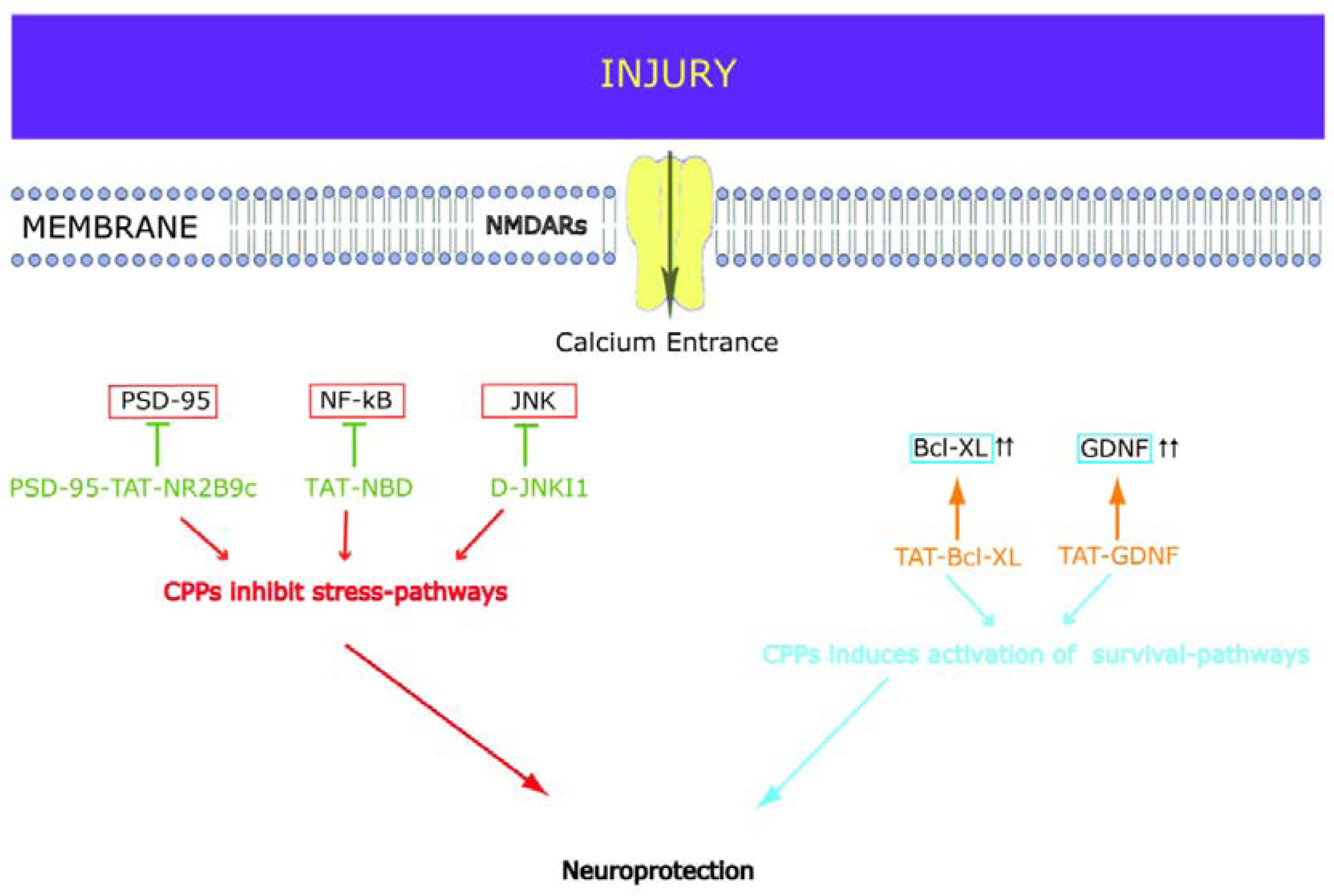
2. Neuroprotective CPPs
2.1. D-JNKI1

2.2. PSD-95-Tat-NR2B9c
2.3. NF-kB
2.4. GDNF
2.5. Bcl-xL
3. CPPs in Chronic Neurodegenerative Disorders
4. Limitations and Pitfalls
4.1. Delivery Strategies: Tissue/Cell Specificity
4.2. Toxicity
4.3. Stability
4.4. Immunogenicity
5. Conclusions
Acknowledgements
References
- Sakurai, H.; Kawabata, K.; Sakurai, F.; Nakagawa, S.; Mizuguchi, H. Innate immune response induced by gene delivery vectors. Int. J. Pharm. 2008, 354, 9–15. [Google Scholar]
- Chiu, Y.L.; Ali, A.; Chu, C.Y.; Cao, H.; Rana, T.M. Visualizing a correlation between siRNA localization, cellular uptake, and RNAi in living cells. Chem. Biol. 2004, 11, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Rothbard, J.B.; Garlington, S.; Lin, Q.; Kirschberg, T.; Kreider, E.; McGrane, P.L.; Wender, P.A.; Khavari, P.A. Conjugation of arginine oligomers to cyclosporin A facilitates topical delivery and inhibition of inflammation. Nat. Med. 2000, 6, 1253–1257. [Google Scholar]
- Lewin, M.; Carlesso, N.; Tung, C.H.; Tang, X.W.; Cory, D.; Scadden, D.T.; Weissleder, R. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat. Biotechnol. 2000, 18, 410–414. [Google Scholar]
- Nguyen, M.; Marcellus, R.C.; Roulston, A.; Watson, M.; Serfass, L.; Murthy Madiraju, S.R.; Goulet, D.; Viallet, J.; Belec, L.; Billot, X.; Acoca, S.; Purisima, E.; Wiegmans, A.; Cluse, L.; Johnstone, R.W.; Beauparlant, P.; Shore, G.C. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 19512–19517. [Google Scholar]
- Park, C.M.; Bruncko, M.; Adickes, J.; Bauch, J.; Ding, H.; Kunzer, A.; Marsh, K.C.; Nimmer, P.; Shoemaker, A.R.; Song, X.; Tahir, S.K.; Tse, C.; Wang, X.; Wendt, M.D.; Yang, X.; Zhang, H.; Fesik, S.W.; Rosenberg, S.H.; Elmore, S.W. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J. Med. Chem. 2008, 51, 6902–6915. [Google Scholar] [PubMed]
- Jarver, P.; Langel, K.; El-Andaloussi, S.; Langel, U. Applications of cell-penetrating peptides in regulation of gene expression. Biochem. Soc. Trans. 2007, 35, 770–774. [Google Scholar]
- Fischer, R.; Fotin-Mleczek, M.; Hufnagel, H.; Brock, R. Break on through to the other side-biophysics and cell biology shed light on cell-penetrating peptides. ChemBioChem 2005, 6, 2126–2142. [Google Scholar]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef] [PubMed]
- Repici, M.; Centeno, C.; Tomasi, S.; Forloni, G.; Bonny, C.; Vercelli, A.; Borsello, T. Time-course of c-Jun N-terminal kinase activation after cerebral ischemia and effect of D-JNKI1 on c-Jun and caspase-3 activation. Neuroscience 2007, 150, 40–49. [Google Scholar]
- Mathupala, S.P. Delivery of small-interfering RNA (siRNA) to the brain. Expert Opin. Ther. Pat. 2009, 19, 137–140. [Google Scholar]
- Szeto, H.H. Development of mitochondria-targeted aromatic-cationic peptides for neurodegenerative diseases. Ann. N.Y. Acad. Sci. 2008, 1147, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar]
- Torchilin, V.P. Tatp-mediated intracellular delivery of pharmaceutical nanocarriers. Biochem. Soc. Trans. 2007, 35, 816–820. [Google Scholar]
- Fischer, P.M. Cellular uptake mechanisms and potential therapeutic utility of peptidic cell delivery vectors: Progress 2001-2006. Med. Res. Rev. 2007, 27, 755–795. [Google Scholar]
- Cardozo, A.K.; Buchillier, V.; Mathieu, M.; Chen, J.; Ortis, F.; Ladriere, L.; Allaman-Pillet, N.; Poirot, O.; Kellenberger, S.; Beckmann, J. S.; Eizirik, D.L.; Bonny, C.; Maurer, F. Cell-permeable peptides induce dose- and length-dependent cytotoxic effects. Biochim. Biophys. Acta 2007, 1768, 2222–2234. [Google Scholar] [CrossRef] [PubMed]
- Foerg, C.; Merkle, H.P. On the biomedical promise of cell penetrating peptides: Limits versus prospects. J. Pharm. Sci. 2008, 97, 144–162. [Google Scholar]
- Said Hassane, F.; Saleh, A.F.; Abes, R.; Gait, M.J.; Lebleu, B. Cell penetrating peptides: Overview and applications to the delivery of oligonucleotides. Cell Mol. Life Sci. 2009. [Google Scholar] [CrossRef]
- Hardingham, G.E. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem. Soc. Trans. 2009, 37, 1147–1160. [Google Scholar]
- Borsello, T.; Forloni, G. JNK signalling: A possible target to prevent neurodegeneration. Curr. Pharm. Des. 2007, 13, 1875–1886. [Google Scholar]
- Benn, S.C.; Woolf, C.J. Adult neuron survival strategies--slamming on the brakes. Nat. Rev. Neurosci. 2004, 5, 686–700. [Google Scholar]
- Fletcher, S.; Hamilton, A.D. Targeting protein-protein interactions by rational design: Mimicry of protein surfaces. J. R. Soc. Interface 2006, 3, 215–233. [Google Scholar]
- Block, P.; Weskamp, N.; Wolf, A.; Klebe, G. Strategies to search and design stabilizers of protein-protein interactions: A feasibility study. Proteins 2007, 68, 170–186. [Google Scholar]
- Bonomi, M.; Gervasio, F.L.; Tiana, G.; Provasi, D.; Broglia, R.A.; Parrinello, M. Insight into the folding inhibition of the HIV-1 protease by a small peptide. Biophys. J. 2007, 93, 2813–2821. [Google Scholar]
- Bonny, C.; Oberson, A.; Negri, S.; Sauser, C.; Schorderet, D.F. Cell-permeable peptide inhibitors of JNK: Novel blockers of beta-cell death. Diabetes 2001, 50, 77–82. [Google Scholar]
- Barr, R.K.; Kendrick, T.S.; Bogoyevitch, M.A. Identification of the critical features of a small peptide inhibitor of JNK activity. J. Biol. Chem. 2002, 277, 10987–10997. [Google Scholar]
- Borsello, T.; Bonny, C. Use of cell-permeable peptides to prevent neuronal degeneration. Trends Mol. Med. 2004, 10, 239–244. [Google Scholar]
- Borsello, T.; Clarke, P.G.; Hirt, L.; Vercelli, A.; Repici, M.; Schorderet, D.F.; Bogousslavsky, J.; Bonny, C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 2003, 9, 1180–1186. [Google Scholar]
- Centeno, C.; Repici, M.; Chatton, J.Y.; Riederer, B.M.; Bonny, C.; Nicod, P.; Price, M.; Clarke, P.G.; Papa, S.; Franzoso, G.; Borsello, T. Role of the JNK pathway in NMDA-mediated excitotoxicity of cortical neurons. Cell Death Differ. 2007, 14, 240–253. [Google Scholar]
- Hirt, L.; Badaut, J.; Thevenet, J.; Granziera, C.; Regli, L.; Maurer, F.; Bonny, C.; Bogousslavsky, J. D-JNKI1, a cell-penetrating c-Jun-N-terminal kinase inhibitor, protects against cell death in severe cerebral ischemia. Stroke 2004, 35, 1738–1743. [Google Scholar] [CrossRef] [PubMed]
- Esneault, E.; Castagne, V.; Moser, P.; Bonny, C.; Bernaudin, M. D-JNKi, a peptide inhibitor of c-Jun N-terminal kinase, promotes functional recovery after transient focal cerebral ischemia in rats. Neuroscience 2008, 152, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Yang, X.; Yang, J.; Wax, M.B. Role of tumor necrosis factor receptor-1 in the death of retinal ganglion cells following optic nerve crush injury in mice. Brain Res. 2004, 996, 202–212. [Google Scholar]
- Wang, J.; Van De Water, T.R.; Bonny, C.; de Ribaupierre, F.; Puel, J.L.; Zine, A. A peptide inhibitor of c-Jun N-terminal kinase protects against both aminoglycoside and acoustic trauma-induced auditory hair cell death and hearing loss. J. Neurosci. 2003, 23, 8596–8607. [Google Scholar]
- Zhuang, Z.Y.; Wen, Y.R.; Zhang, D.R.; Borsello, T.; Bonny, C.; Strichartz, G.R.; Decosterd, I.; Ji, R.R. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: Respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J. Neurosci. 2006, 26, 3551–3560. [Google Scholar]
- Beckham, J.D.; Goody, R.J.; Clarke, P.; Bonny, C.; Tyler, K.L. Novel strategy for treatment of viral central nervous system infection by using a cell-permeating inhibitor of c-Jun N-terminal kinase. J. Virol. 2007, 81, 6984–6992. [Google Scholar]
- Hui, L.; Zatloukal, K.; Scheuch, H.; Stepniak, E.; Wagner, E.F. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J. Clin. Invest. 2008, 118, 3943–3953. [Google Scholar]
- Lehnert, M.; Relja, B.; Sun-Young Lee, V.; Schwestka, B.; Henrich, D.; Czerny, C.; Froh, M.; Borsello, T.; Marzi, I. A peptide inhibitor of C-jun N-terminal kinase modulates hepatic damage and the inflammatory response after hemorrhagic shock and resuscitation. Shock 2008, 30, 159–165. [Google Scholar]
- Aarts, M.; Liu, Y.; Liu, L.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.T.; Salter, M.W.; Tymianski, M. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science 2002, 298, 846–850. [Google Scholar]
- Sun, H.S.; Doucette, T.A.; Liu, Y.; Fang, Y.; Teves, L.; Aarts, M.; Ryan, C.L.; Bernard, P.B.; Lau, A.; Forder, J.P.; Salter, M.W.; Wang, Y.T.; Tasker, R.A.; Tymianski, M. Effectiveness of PSD95 inhibitors in permanent and transient focal ischemia in the rat. Stroke 2008, 39, 2544–2553. [Google Scholar]
- Soriano, F.X.; Martel, M.A.; Papadia, S.; Vaslin, A.; Baxter, P.; Rickman, C.; Forder, J.; Tymianski, M.; Duncan, R.; Aarts, M.; Clarke, P.; Wyllie, D.J.; Hardingham, G.E. Specific targeting of pro-death NMDA receptor signals with differing reliance on the NR2B PDZ ligand. J. Neurosci. 2008, 28, 10696–10710. [Google Scholar]
- Dykstra, C.M.; Ratnam, M.; Gurd, J.W. Neuroprotection after status epilepticus by targeting protein interactions with postsynaptic density protein 95. J. Neuropathol. Exp. Neurol. 2009, 68, 823–831. [Google Scholar]
- Tao, F.; Su, Q.; Johns, R.A. Cell-permeable peptide Tat-PSD-95 PDZ2 inhibits chronic inflammatory pain behaviors in mice. Mol. Ther. 2008, 16, 1776–1782. [Google Scholar]
- Sarnico, I.; Lanzillotta, A.; Benarese, M.; Alghisi, M.; Baiguera, C.; Battistin, L.; Spano, P.; Pizzi, M. NF-kappaB dimers in the regulation of neuronal survival. Int. Rev. Neurobiol. 2009, 85, 351–362. [Google Scholar]
- Nijboer, C.H.; Heijnen, C.J.; Groenendaal, F.; May, M.J.; van Bel, F.; Kavelaars, A. Strong neuroprotection by inhibition of NF-kappaB after neonatal hypoxia-ischemia involves apoptotic mechanisms but is independent of cytokines. Stroke 2008, 39, 2129–2137. [Google Scholar]
- Nijboer, C.H.; Heijnen, C.J.; Groenendaal, F.; May, M.J.; van Bel, F.; Kavelaars, A. A dual role of the NF-kappaB pathway in neonatal hypoxic-ischemic brain damage. Stroke 2008, 39, 2578–2586. [Google Scholar]
- Orange, J.S.; May, M.J. Cell penetrating peptide inhibitors of nuclear factor-kappa B. Cell Mol. Life Sci. 2008, 65, 3564–3591. [Google Scholar]
- Kilic, U.; Kilic, E.; Dietz, G.P.; Bahr, M. Intravenous TAT-GDNF is protective after focal cerebral ischemia in mice. Stroke 2003, 34, 1304–1310. [Google Scholar]
- Kilic, U.; Kilic, E.; Dietz, G.P.; Bahr, M. The TAT protein transduction domain enhances the neuroprotective effect of glial-cell-line-derived neurotrophic factor after optic nerve transection. Neurodegener Dis. 2004, 1, 44–49. [Google Scholar]
- Hu, Y.; Benedict, M.A.; Wu, D.; Inohara, N.; Nunez, G. Bcl-XL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc. Natl. Acad. Sci. USA 1998, 95, 4386–4391. [Google Scholar]
- Gillardon, F.; Lenz, C.; Waschke, K.F.; Krajewski, S.; Reed, J.C.; Zimmermann, M.; Kuschinsky, W. Altered expression of Bcl-2, Bcl-X, Bax, and c-Fos colocalizes with DNA fragmentation and ischemic cell damage following middle cerebral artery occlusion in rats. Brain Res. Mol. Brain Res. 1996, 40, 254–260. [Google Scholar] [PubMed]
- Isenmann, S.; Stoll, G.; Schroeter, M.; Krajewski, S.; Reed, J.C.; Bahr, M. Differential regulation of Bax, Bcl-2, and Bcl-X proteins in focal cortical ischemia in the rat. Brain Pathol. 1998, 8, 49–62. [Google Scholar] [PubMed]
- Cao, G.; Pei, W.; Ge, H.; Liang, Q.; Luo, Y.; Sharp, F.R.; Lu, A.; Ran, R.; Graham, S.H.; Chen, J. In Vivo Delivery of a Bcl-xL Fusion Protein Containing the TAT Protein Transduction Domain Protects against Ischemic Brain Injury and Neuronal Apoptosis. J. Neurosci. 2002, 22, 5423–5431. [Google Scholar] [PubMed]
- Kilic, E.; Dietz, G.P.; Hermann, D.M.; Bahr, M. Intravenous TAT-Bcl-Xl is protective after middle cerebral artery occlusion in mice. Ann. Neurol. 2002, 52, 617–622. [Google Scholar]
- Kilic, E.; Kilic, U.; Hermann, D.M. TAT fusion proteins against ischemic stroke: Current status and future perspectives. Front Biosci. 2006, 11, 1716–1721. [Google Scholar]
- Dietz, G.P.; Stockhausen, K.V.; Dietz, B.; Falkenburger, B.H.; Valbuena, P.; Opazo, F.; Lingor, P.; Meuer, K.; Weishaupt, J.H.; Schulz, J.B.; Bahr, M. Membrane-permeable Bcl-xL prevents MPTP-induced dopaminergic neuronal loss in the substantia nigra. J. Neurochem. 2008, 104, 757–765. [Google Scholar]
- Jia, H.; Lohr, M.; Jezequel, S.; Davis, D.; Shaikh, S.; Selwood, D.; Zachary, I. Cysteine-rich and basic domain HIV-1 Tat peptides inhibit angiogenesis and induce endothelial cell apoptosis. Biochem. Biophys. Res. Commun. 2001, 283, 469–479. [Google Scholar]
- Fu, A.L.; Li, Q.; Dong, Z.H.; Huang, S.J.; Wang, Y.X.; Sun, M.J. Alternative therapy of Alzheimer's disease via supplementation with choline acetyltransferase. Neurosci. Lett 2004, 368, 258–262. [Google Scholar]
- Yeon, S.W.; Jeon, Y.J.; Hwang, E.M.; Kim, T.Y. Effects of peptides derived from BACE1 catalytic domain on APP processing. Peptides 2007, 28, 838–844. [Google Scholar]
- Dasgupta, S.; Jana, M.; Zhou, Y.; Fung, Y.K.; Ghosh, S.; Pahan, K. Antineuroinflammatory effect of NF-kappaB essential modifier-binding domain peptides in the adoptive transfer model of experimental allergic encephalomyelitis. J. Immunol. 2004, 173, 1344–1354. [Google Scholar]
- Szeto, H.H. Mitochondria-targeted peptide antioxidants: Novel neuroprotective agents. Aaps. J. 2006, 8, 521–531. [Google Scholar]
- Wagner, E.; Culmsee, C.; Boeckle, S. Targeting of polyplexes: Toward synthetic virus vector systems. Adv. Genet. 2005, 53, 333–354. [Google Scholar]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar]
- Repici, M.; Mare, L.; Colombo, A.; Ploia, C.; Sclip, A.; Bonny, C.; Nicod, P.; Salmona, M.; Borsello, T. c-Jun N-terminal kinase binding domain-dependent phosphorylation of mitogen-activated protein kinase kinase 4 and mitogen-activated protein kinase kinase 7 and balancing cross-talk between c-Jun N-terminal kinase and extracellular signal-regulated kinase pathways in cortical neurons. Neuroscience 2009, 159, 94–103. [Google Scholar]
- Vives, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar]
- Trehin, R.; Merkle, H.P. Chances and pitfalls of cell penetrating peptides for cellular drug delivery. Eur. J. Pharm. Biopharm. 2004, 58, 209–223. [Google Scholar]
- Drin, G.; Cottin, S.; Blanc, E.; Rees, A.R.; Temsamani, J. Studies on the internalization mechanism of cationic cell-penetrating peptides. J. Biol. Chem. 2003, 278, 31192–31201. [Google Scholar]
- Lindgren, M.E.; Hallbrink, M.M.; Elmquist, A.M.; Langel, U. Passage of cell-penetrating peptides across a human epithelial cell layer in vitro. Biochem. J. 2004, 377, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Koppelhus, U.; Awasthi, S.K.; Zachar, V.; Holst, H.U.; Ebbesen, P.; Nielsen, P.E. Cell-dependent differential cellular uptake of PNA, peptides, and PNA-peptide conjugates. Antisense Nucleic Acid Drug Dev. 2002, 12, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Brugidou, J.; Legrand, C.; Mery, J.; Rabie, A. The retro-inverso form of a homeobox-derived short peptide is rapidly internalised by cultured neurones: A new basis for an efficient intracellular delivery system. Biochem. Biophys. Res. Commun. 1995, 214, 685–693. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Antoniou, X.; Borsello, T. Cell Permeable Peptides: A Promising Tool to Deliver Neuroprotective Agents in the Brain. Pharmaceuticals 2010, 3, 379-392. https://doi.org/10.3390/ph3020379
Antoniou X, Borsello T. Cell Permeable Peptides: A Promising Tool to Deliver Neuroprotective Agents in the Brain. Pharmaceuticals. 2010; 3(2):379-392. https://doi.org/10.3390/ph3020379
Chicago/Turabian StyleAntoniou, Xanthi, and Tiziana Borsello. 2010. "Cell Permeable Peptides: A Promising Tool to Deliver Neuroprotective Agents in the Brain" Pharmaceuticals 3, no. 2: 379-392. https://doi.org/10.3390/ph3020379