

Therapeutic Implications of Ceritinib in Cholangiocarcinoma beyond ALK Expression and Mutation
 ,
,  ,
,  , , ,
, , ,  , , and add
Show full author list
, , and add
Show full author list
Abstract
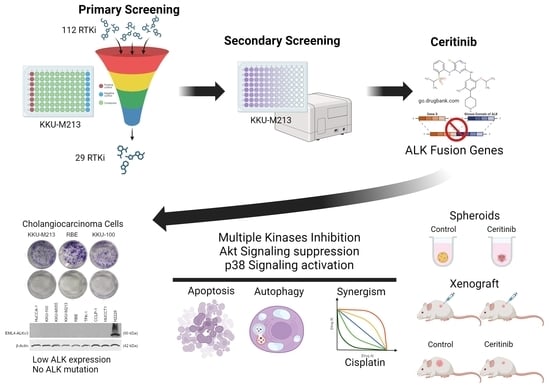
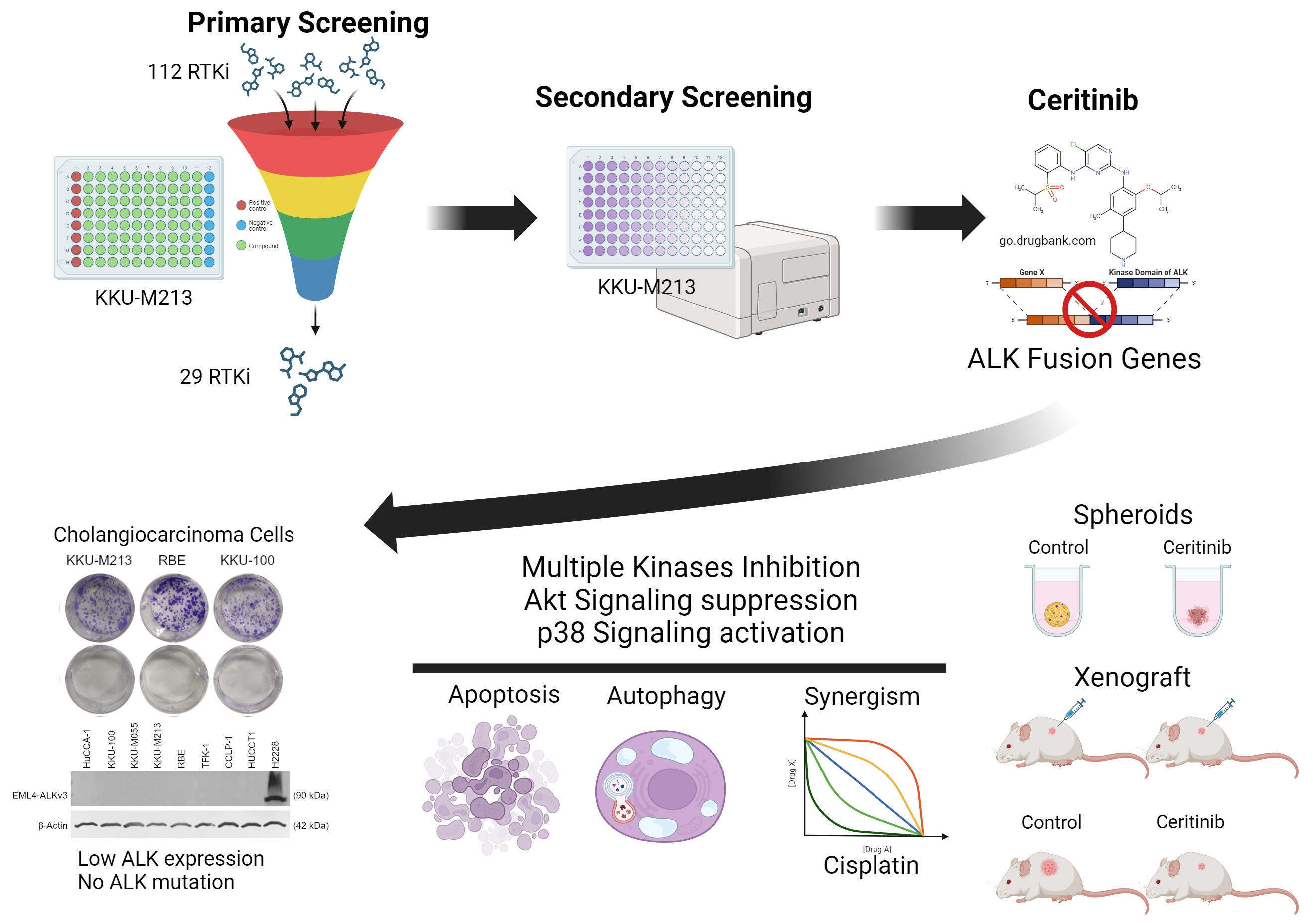
:
1. Introduction
2. Results
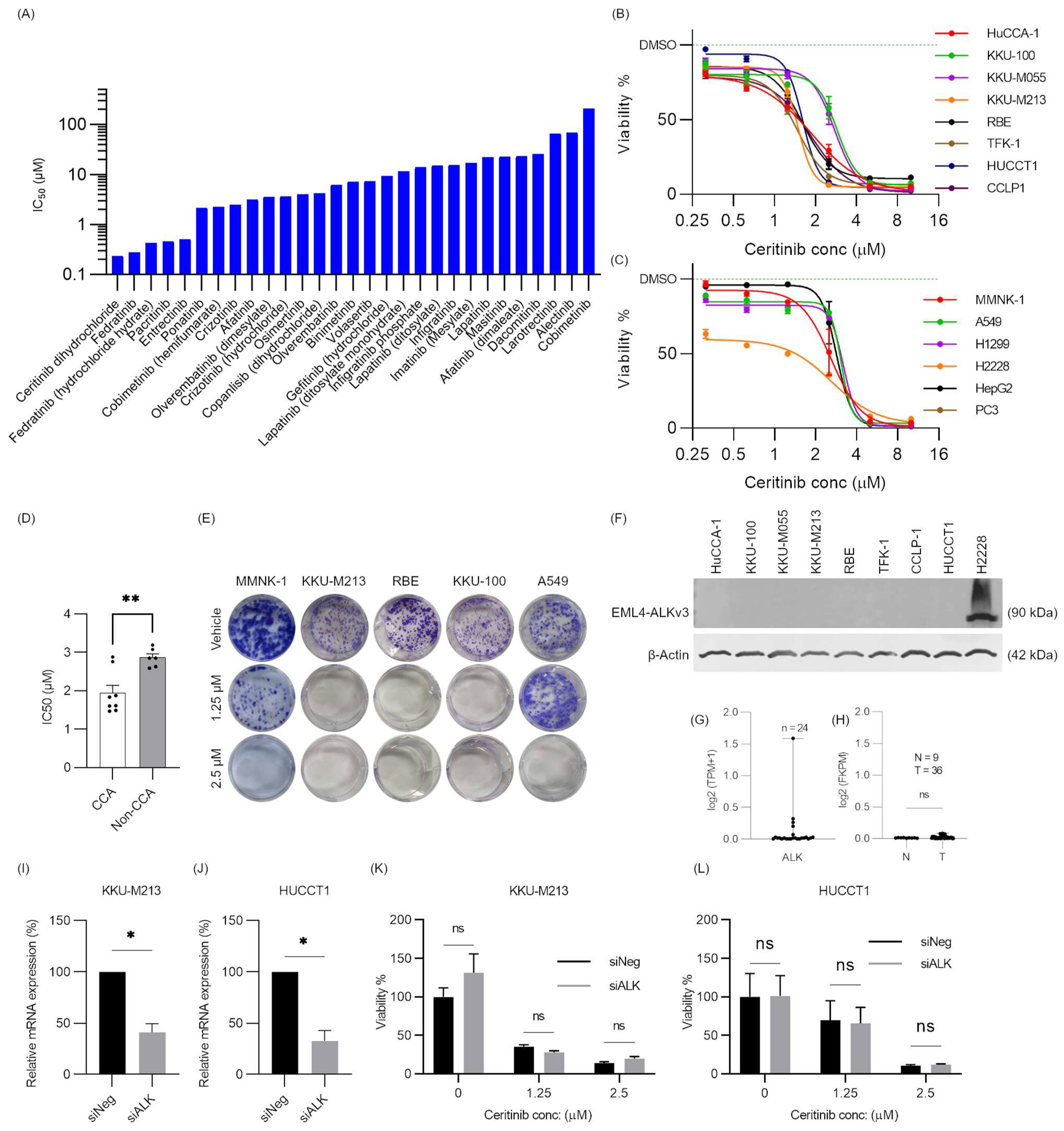
2.1. Receptor Tyrosine Kinase Inhibitor Screening in CCA
2.2. Ceritinib Is Particularly Cytotoxic to CCA Cells
2.3. Ceritinib-Induced Cytotoxicity in CCA Cells Is ALK Mutation/Expression Independent
2.4. Prediction of Ceritinib-Induced Cell Death Mechanism and Signaling Pathways
2.5. Ceritinib-Induced Apoptosis and Autophagy in CCA
2.6. Ceritinib Alters Multiple Signaling Pathways of CCA Cells
2.7. Ceritinib and Cisplatin Combination Showed Synergistic Activity
2.8. Ceritinib Showed Potent Cytotoxic Effects in Spheroid Models and Xenograft Models
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Cultures
4.2. Chemical and Reagents
4.3. Cell Viability Assays
4.4. Colony Formation Assays
4.5. Annexin V and PI Staining for FACS
4.6. Datamining of ALK Gene Expression and Mutation Status in CCA
4.7. siRNA-Mediated Gene Silencing
4.8. qRT-PCR
4.9. SDS-PAGE and Immunoblotting
4.10. Immuno-Fluorescence and Fluorescent Microscopy
4.11. Autophagy Flux Study
4.12. Antibody Array
4.13. Drug Synergism Study
4.14. Spheroid Culture
4.15. Xenograft Model
4.15.1. Preparing Cells for Transplantation
4.15.2. Subcutaneous Injection of Cells and Treatment
4.16. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blechacz, B. Cholangiocarcinoma: Current Knowledge and New Developments. Gut Liver 2017, 11, 13–26. [Google Scholar] [CrossRef]
- DeOliveira, M.L.; Cunningham, S.C.; Cameron, J.L.; Kamangar, F.; Winter, J.M.; Lillemoe, K.D.; Choti, M.A.; Yeo, C.J.; Schulick, R.D. Cholangiocarcinoma: Thirty-one-year experience with 564 patients at a single institution. Ann. Surg. 2007, 245, 755–762. [Google Scholar] [CrossRef]
- Nakeeb, A.; Pitt, H.A.; Sohn, T.A.; Coleman, J.; Abrams, R.A.; Piantadosi, S.; Hruban, R.H.; Lillemoe, K.D.; Yeo, C.J.; Cameron, J.L. Cholangiocarcinoma. A spectrum of intrahepatic, perihilar, and distal tumors. Ann. Surg. 1996, 224, 463–473, discussion 473–465. [Google Scholar] [CrossRef]
- Khan, S.A.; Thomas, H.C.; Davidson, B.R.; Taylor-Robinson, S.D. Cholangiocarcinoma. Lancet 2005, 366, 1303–1314. [Google Scholar] [CrossRef]
- Mosconi, S.; Beretta, G.D.; Labianca, R.; Zampino, M.G.; Gatta, G.; Heinemann, V. Cholangiocarcinoma. Crit. Rev. Oncol. Hematol. 2009, 69, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Treeprasertsuk, S.; Poovorawan, K.; Soonthornworasiri, N.; Chaiteerakij, R.; Thanapirom, K.; Mairiang, P.; Sawadpanich, K.; Sonsiri, K.; Mahachai, V.; Phaosawasdi, K. A significant cancer burden and high mortality of intrahepatic cholangiocarcinoma in Thailand: A nationwide database study. BMC Gastroenterol. 2017, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Patel, T. Cholangiocarcinoma–controversies and challenges. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Vidili, G.; Rengo, M.; Bujanda, L.; Ponz-Sarvise, M.; Lamarca, A. Clinical presentation, diagnosis and staging of cholangiocarcinoma. Liver Int. 2019, 39 (Suppl. S1), 98–107. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef]
- Eckmann, K.R.; Patel, D.K.; Landgraf, A.; Slade, J.H.; Lin, E.; Kaur, H.; Loyer, E.; Weatherly, J.M.; Javle, M. Chemotherapy outcomes for the treatment of unresectable intrahepatic and hilar cholangiocarcinoma: A retrospective analysis. Gastrointest. Cancer Res. 2011, 4, 155–160. [Google Scholar]
- Patel, T.; Singh, P. Cholangiocarcinoma: Emerging approaches to a challenging cancer. Curr. Opin. Gastroenterol. 2007, 23, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Squires, M.H.; Cloyd, J.M.; Dillhoff, M.; Schmidt, C.; Pawlik, T.M. Challenges of surgical management of intrahepatic cholangiocarcinoma. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J.G.; Lozano, E.; Herraez, E.; Asensio, M.; Di Giacomo, S.; Romero, M.R.; Briz, O.; Serrano, M.A.; Efferth, T.; Macias, R.I.R. Chemoresistance and chemosensitization in cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1444–1453. [Google Scholar] [CrossRef]
- Rahnemai-Azar, A.A.; Abbasi, A.; Acher, A.W.; Weber, S.M.; Pawlik, T.M. Emerging pathways for precision medicine in management of cholangiocarcinoma. Surg. Oncol. 2020, 35, 47–55. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Friboulet, L.; Li, N.; Katayama, R.; Lee, C.C.; Gainor, J.F.; Crystal, A.S.; Michellys, P.Y.; Awad, M.M.; Yanagitani, N.; Kim, S.; et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014, 4, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Tan, D.S.W.; Chiari, R.; Wu, Y.L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): A randomised, open-label, phase 3 study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Holla, V.R.; Elamin, Y.Y.; Bailey, A.M.; Johnson, A.M.; Litzenburger, B.C.; Khotskaya, Y.B.; Sanchez, N.S.; Zeng, J.; Shufean, M.A.; Shaw, K.R.; et al. ALK: A tyrosine kinase target for cancer therapy. Cold Spring Harb. Mol. Case Stud. 2017, 3, a001115. [Google Scholar] [CrossRef]
- Yu, Z.; Zhao, R. Inhibition of anaplastic lymphoma kinase promotes apoptosis and suppresses proliferation in human hepatocellular carcinoma. Anticancer Drugs 2018, 29, 513–519. [Google Scholar] [CrossRef]
- Das, A.; Alshareef, M.; Porto, G.B.F.; Infinger, L.K.; Vandergrift, W.A., 3rd; Lindhorst, S.M.; Varma, A.K.; Patel, S.J.; Cachia, D. Preconditioning with INC280 and LDK378 drugs sensitizes MGMT-unmethylated glioblastoma to temozolomide: Pre-clinical assessment. J. Neurol. Sci. 2020, 418, 117102. [Google Scholar] [CrossRef] [PubMed]
- Jamshed, M.B.; Munir, F.; Shahid, N.; Sadiq, U.; Muhammad, S.A.; Ghanem, N.B.; Zhong, H.; Li, X.; Zhang, Q. Antitumor activity and combined inhibitory effect of ceritinib with gemcitabine in pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G109–G119. [Google Scholar] [CrossRef] [PubMed]
- Myint, K.Z.; Sueca-Comes, M.; Collier, P.; Balasubramanian, B.; Venkatraman, S.; Gordan, J.; Zaitoun, A.M.; Mukherjee, A.; Arora, A.; Larbcharoensub, N.; et al. Preclinical evidence for anaplastic lymphoma kinase inhibitors as novel therapeutic treatments for cholangiocarcinoma. Front. Oncol. 2023, 13, 1184900. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Murakami, H.; Horiike, A.; Takahashi, T.; Hirai, F.; Suenaga, N.; Tajima, T.; Tokushige, K.; Ishii, M.; Boral, A.; et al. Phase I Study of Ceritinib (LDK378) in Japanese Patients with Advanced, Anaplastic Lymphoma Kinase-Rearranged Non-Small-Cell Lung Cancer or Other Tumors. J. Thorac. Oncol. 2015, 10, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 1189–1197. [Google Scholar] [CrossRef]
- Marsilje, T.H.; Pei, W.; Chen, B.; Lu, W.; Uno, T.; Jin, Y.; Jiang, T.; Kim, S.; Li, N.; Warmuth, M.; et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulf onyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J. Med. Chem. 2013, 56, 5675–5690. [Google Scholar] [CrossRef]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Vewinger, N.; Huprich, S.; Seidmann, L.; Russo, A.; Alt, F.; Bender, H.; Sommer, C.; Samuel, D.; Lehmann, N.; Backes, N.; et al. IGF1R Is a Potential New Therapeutic Target for HGNET-BCOR Brain Tumor Patients. Int. J. Mol. Sci. 2019, 20, 3027. [Google Scholar] [CrossRef]
- Wang, F.; Bank, T.; Malnassy, G.; Arteaga, M.; Shang, N.; Dalheim, A.; Ding, X.; Cotler, S.J.; Denning, M.F.; Nishimura, M.I.; et al. Inhibition of insulin-like growth factor 1 receptor enhances the efficacy of sorafenib in inhibiting hepatocellular carcinoma cell growth and survival. Hepatol. Commun. 2018, 2, 732–746. [Google Scholar] [CrossRef]
- Kuenzi, B.M.; Remsing Rix, L.L.; Stewart, P.A.; Fang, B.; Kinose, F.; Bryant, A.T.; Boyle, T.A.; Koomen, J.M.; Haura, E.B.; Rix, U. Polypharmacology-based ceritinib repurposing using integrated functional proteomics. Nat. Chem. Biol. 2017, 13, 1222–1231. [Google Scholar] [CrossRef]
- Chang, H.; Zou, Z. Targeting autophagy to overcome drug resistance: Further developments. J. Hematol. Oncol. 2020, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, L.L.; Shen, L.; Xu, X.M.; Yu, H.G. Regulation of AKT gene expression by cisplatin. Oncol. Lett. 2013, 5, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.J.; Wang, J.; Zhou, J.Y.; Wu, G.S. Role of the Akt/mTOR survival pathway in cisplatin resistance in ovarian cancer cells. Biochem. Biophys. Res. Commun. 2010, 394, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bao, C.; Mu, Q.; Chen, J.; Wang, J.; Mi, Y.; Sayari, A.J.; Chen, Y.; Guo, M. Reversal of cisplatin resistance by inhibiting PI3K/Akt signal pathway in human lung cancer cells. Neoplasma 2016, 63, 362–370. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.D.; Pinson, C.W.; Berlin, J.; Chari, R.S. Diagnosis and treatment of cholangiocarcinoma. Oncologist 2004, 9, 43–57. [Google Scholar] [CrossRef]
- Ramirez-Merino, N.; Aix, S.P.; Cortes-Funes, H. Chemotherapy for cholangiocarcinoma: An update. World J. Gastrointest. Oncol. 2013, 5, 171–176. [Google Scholar] [CrossRef]
- Simile, M.M.; Bagella, P.; Vidili, G.; Spanu, A.; Manetti, R.; Seddaiu, M.A.; Babudieri, S.; Madeddu, G.; Serra, P.A.; Altana, M.; et al. Targeted Therapies in Cholangiocarcinoma: Emerging Evidence from Clinical Trials. Medicina 2019, 55, 42. [Google Scholar] [CrossRef]
- Chiang, N.J.; Hsu, C.; Chen, J.S.; Tsou, H.H.; Shen, Y.Y.; Chao, Y.; Chen, M.H.; Yeh, T.S.; Shan, Y.S.; Huang, S.F.; et al. Expression levels of ROS1/ALK/c-MET and therapeutic efficacy of cetuximab plus chemotherapy in advanced biliary tract cancer. Sci. Rep. 2016, 6, 25369. [Google Scholar] [CrossRef]
- Augustin, J.; Gabignon, C.; Scriva, A.; Menu, L.; Calmel, C.; Scatton, O.; Paye, F.; Fléjou, J.F.; Praz, F.; Cervera, P.; et al. Testing for ROS1, ALK, MET, and HER2 rearrangements and amplifications in a large series of biliary tract adenocarcinomas. Virchows Arch. Int. J. Pathol. 2020, 477, 33–45. [Google Scholar] [CrossRef]
- Zarin, D.A.; Fain, K.M.; Dobbins, H.D.; Tse, T.; Williams, R.J. 10-Year Update on Study Results Submitted to ClinicalTrials.gov. N. Engl. J. Med. 2019, 381, 1966–1974. [Google Scholar] [CrossRef]
- Metz, J.T.; Hajduk, P.J. Rational approaches to targeted polypharmacology: Creating and navigating protein-ligand interaction networks. Curr. Opin. Chem. Biol. 2010, 14, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.S.; Gupta, R.; Liguori, M.J.; Hu, M.; Huang, X.; Mantena, S.R.; Mittelstadt, S.W.; Blomme, E.A.G.; Van Vleet, T.R. Novel Computational Approach to Predict Off-Target Interactions for Small Molecules. Front. Big Data 2019, 2, 25. [Google Scholar] [CrossRef] [PubMed]
- Peon, A.; Naulaerts, S.; Ballester, P.J. Predicting the Reliability of Drug-target Interaction Predictions with Maximum Coverage of Target Space. Sci. Rep. 2017, 7, 3820. [Google Scholar] [CrossRef] [PubMed]
- Krug, M.; Hilgeroth, A. Recent advances in the development of multi-kinase inhibitors. Mini Rev. Med. Chem. 2008, 8, 1312–1327. [Google Scholar] [CrossRef] [PubMed]
- Broekman, F.; Giovannetti, E.; Peters, G.J. Tyrosine kinase inhibitors: Multi-targeted or single-targeted? World J. Clin. Oncol. 2011, 2, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Tian, A.-L.; Durand, S.; Aprahamian, F.; Nirmalathasan, N.; Xie, W.; Liu, P.; Zhao, L.; Zhang, S.; Pan, H.; et al. Isobacachalcone induces autophagy and improves the outcome of immunogenic chemotherapy. Cell Death Dis. 2020, 11, 1015. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.L. Regulation of autophagy by p38α MAPK. Autophagy 2010, 6, 292–293. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.-H. AKT as a Therapeutic Target for Cancer. J. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Yothaisong, S.; Dokduang, H.; Techasen, A.; Namwat, N.; Yongvanit, P.; Bhudhisawasdi, V.; Puapairoj, A.; Riggins, G.J.; Loilome, W. Increased activation of PI3K/AKT signaling pathway is associated with cholangiocarcinoma metastasis and PI3K/mTOR inhibition presents a possible therapeutic strategy. Tumor Biol. 2013, 34, 3637–3648. [Google Scholar] [CrossRef]
- Wilson, J.M.; Kunnimalaiyaan, S.; Kunnimalaiyaan, M.; Gamblin, T.C. Inhibition of the AKT pathway in cholangiocarcinoma by MK2206 reduces cellular viability via induction of apoptosis. Cancer Cell Int. 2015, 15, 13. [Google Scholar] [CrossRef]
- Wu, T.; Leng, J.; Han, C.; Demetris, A.J. The cyclooxygenase-2 inhibitor celecoxib blocks phosphorylation of Akt and induces apoptosis in human cholangiocarcinoma cells. Mol. Cancer Ther. 2004, 3, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Vinci, M.; Gowan, S.; Boxall, F.; Patterson, L.; Zimmermann, M.; Court, W.; Lomas, C.; Mendiola, M.; Hardisson, D.; Eccles, S.A. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012, 10, 29. [Google Scholar] [CrossRef]
- Ravi, M.; Paramesh, V.; Kaviya, S.R.; Anuradha, E.; Solomon, F.D. 3D cell culture systems: Advantages and applications. J. Cell. Physiol. 2015, 230, 16–26. [Google Scholar] [CrossRef]
- Kimlin, L.C.; Casagrande, G.; Virador, V.M. In vitro three-dimensional (3D) models in cancer research: An update. Mol. Carcinog. 2013, 52, 167–182. [Google Scholar] [CrossRef]
- Shehzad, A.; Ravinayagam, V.; AlRumaih, H.; Aljafary, M.; Almohazey, D.; Almofty, S.; Al-Rashid, N.A.; Al-Suhaimi, E.A. Application of Three-dimensional (3D) Tumor Cell Culture Systems and Mechanism of Drug Resistance. Curr. Pharm. Des. 2019, 25, 3599–3607. [Google Scholar] [CrossRef]
- Lee, M.S.; Jung, K.; Song, J.Y.; Sung, M.J.; Ahn, S.B.; Lee, B.; Oh, D.Y.; Choi, Y.L. IRS2 Amplification as a Predictive Biomarker in Response to Ceritinib in Small Cell Lung Cancer. Mol. Ther. Oncolytics 2020, 16, 188–196. [Google Scholar] [CrossRef]
- Fountzilas, C.; Adjei, A.; Opyrchal, M.; Evans, R.; Ghasemi, M.; Attwood, K.; Groman, A.; Bshara, W.; Goey, A.; Wilton, J.; et al. A phase I study of the anaplastic lymphoma kinase inhibitor ceritinib in combination with gemcitabine-based chemotherapy in patients with advanced solid tumors. Int. J. Cancer 2021, 149, 2063–2074. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Rodig, S.J.; Chirieac, L.R.; Janne, P.A. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur. J. Cancer 2010, 46, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Vander Velde, R.; Yoon, N.; Marusyk, V.; Durmaz, A.; Dhawan, A.; Miroshnychenko, D.; Lozano-Peral, D.; Desai, B.; Balynska, O.; Poleszhuk, J.; et al. Resistance to targeted therapies as a multifactorial, gradual adaptation to inhibitor specific selective pressures. Nat. Commun. 2020, 11, 2393. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.; Creighton, C.J.; Davis, C.; Donehower, L.; Drummond, J.; Wheeler, D.; Ally, A.; Balasundaram, M.; Birol, I.; Butterfield, Y.S.N.; et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Ogawa, S.; Takita, J.; Sanada, M.; Hayashi, Y. Oncogenic mutations of ALK in neuroblastoma. Cancer Sci. 2011, 102, 302–308. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Singsuksawat, E.; Thuwajit, C.; Charngkaew, K.; Thuwajit, P. Increased ETV4 expression correlates with estrogen-enhanced proliferation and invasiveness of cholangiocarcinoma cells. Cancer Cell Int. 2018, 18, 25. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell-Line | Cell Type | Identifier |
|---|---|---|
| MMNK-1 | Immortalized Cholangiocytes | JCRB1554 |
| HuCCA-1 | Cholangiocarcinoma | JCRB1657 |
| KKU-M055 | Cholangiocarcinoma | JCRB1551 |
| KKU-100 | Cholangiocarcinoma | JCRB1568 |
| KKU-M213 | Cholangiocarcinoma | JCRB1557 |
| RBE | Cholangiocarcinoma | RCB1292 |
| TFK-1 | Cholangiocarcinoma | RCB2537 |
| HuCCT1 | Cholangiocarcinoma | JCRB0425 |
| CCLP1 | Cholangiocarcinoma | Cellosaurus RRID: CVCL_0205 |
| HepG2 | Liver Cancer | ATCC HP-8065 |
| A549 | Non-small cell lung cancer | ATCC CCL-185 |
| H2228 | Lung Cancer | ATCC CRL-5935 |
| H1299 | Lung Cancer | ATCC CRL-5803 |
| PC3 | Prostate Cancer | ATCC CRL-1435 |
| Antibody | Type | Organism | Company | Dilution |
|---|---|---|---|---|
| ALK (D5F3) 3633T | Monoclonal | Rabbit | Cell Signaling Technology®, Beverly, MA, USA | 1:1000 |
| Phospho-ALK (Tyr1604) #3341 | Monoclonal | Rabbit | Cell Signaling Technology®, Beverly, MA, USA | 1:1000 |
| Phospho-Akt (Ser473) (193H12) #4058 | Monoclonal | Rabbit | Cell Signaling Technology®, Beverly, MA, USA | 1:1000 |
| Total Akt (BDI111) sc-56878 | Monoclonal | Mouse | Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA | 1:1000 |
| Phospho-p38 MAPK (Thr180/Tyr182) #9211 | Monoclonal | Rabbit | Cell Signaling Technology®, Beverly, MA, USA | 1:1000 |
| p38 MAPK #9212 | Monoclonal | Mouse | Cell Signaling Technology®, Beverly, MA, USA | 1:1000 |
| GAPDH (0411) sc-47724 | Monoclonal | Mouse | Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA | 1:2000 |
| β-actin A1978 | Monoclonal | Mouse | Sigma Aldrich | 1:10,000 |
| PARP #9542 | Monoclonal | Rabbit | Cell Signaling Technology®, Beverly, MA, USA | 1:1000 |
| Cleaved PARP (Asp214) (D64E10) XP® #5625 | Monoclonal | Rabbit | Cell Signaling Technology®, Beverly, MA, USA | 1:1000 |
| Caspase-3 Antibody #9662 | Monoclonal | Rabbit | Cell Signaling Technology®, Beverly, MA, USA | 1:1000 |
| Cleaved Caspase-3 (Asp175) (5A1E) #9664 | Monoclonal | Rabbit | Cell Signaling Technology®, Beverly, MA, USA | 1:1000 |
| LC3B #2775 | Monoclonal | Rabbit | Cell Signaling Technology®, Beverly, MA, USA | 1:1000 |
| Anti-SQSTM1/p62 ab56416 | Monoclonal | Mouse | Abcam, Cambridge, MA, USA | 1:1000 |
| Phospho-mTOR (Ser2448) (D9C2) XP® #5536 | Monoclonal | Rabbit | Cell Signaling Technology®, Beverly, MA, USA | 1:1000 |
| Phospho-4E-BP1 (Thr37/46) (236B4) #2855 | Monoclonal | Rabbit | Cell Signaling Technology®, Beverly, MA, USA | 1:1000 |
| Anti-rabbit IgG, HRP-linked Antibody #7074 | Polyclonal | - | Cell Signaling Technology®, Beverly, MA, USA | 1:1000 |
| Anti-mouse IgG, HRP-linked Antibody #7076 | Polyclonal | - | Cell Signaling Technology®, Beverly, MA, USA | 1:1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Myint, K.Z.; Balasubramanian, B.; Venkatraman, S.; Phimsen, S.; Sripramote, S.; Jantra, J.; Choeiphuk, C.; Mingphruedhi, S.; Muangkaew, P.; Rungsakulkij, N.; et al. Therapeutic Implications of Ceritinib in Cholangiocarcinoma beyond ALK Expression and Mutation. Pharmaceuticals 2024, 17, 197. https://doi.org/10.3390/ph17020197
Myint KZ, Balasubramanian B, Venkatraman S, Phimsen S, Sripramote S, Jantra J, Choeiphuk C, Mingphruedhi S, Muangkaew P, Rungsakulkij N, et al. Therapeutic Implications of Ceritinib in Cholangiocarcinoma beyond ALK Expression and Mutation. Pharmaceuticals. 2024; 17(2):197. https://doi.org/10.3390/ph17020197
Chicago/Turabian StyleMyint, Kyaw Zwar, Brinda Balasubramanian, Simran Venkatraman, Suchada Phimsen, Supisara Sripramote, Jeranan Jantra, Chaiwat Choeiphuk, Somkit Mingphruedhi, Paramin Muangkaew, Narongsak Rungsakulkij, and et al. 2024. "Therapeutic Implications of Ceritinib in Cholangiocarcinoma beyond ALK Expression and Mutation" Pharmaceuticals 17, no. 2: 197. https://doi.org/10.3390/ph17020197